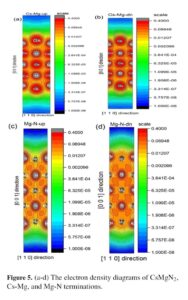
Structural, electronic, and thermoelectric properties of the CsMgN2 compound with its thin film films of Cs-Mg and Mg-N terminations have been studied in a First-Principles study. The total energy changes (E-V) versus the unit cell volume of bulk show better stability in the ferromagnetic than the non-magnetic phase. The E-V diagrams of film Cs-Mg and […]
We have performed density functional theory calculations to study the adsorption of Li, B, N, and O clusters on graphene to clarify the interaction of these atoms with a graphene sheet. The stable structure, the adsorption energy, and the density of states (DOS) of atom-graphene systems are calculated. The obtained results of the structural, adsorption […]
AbstractUsing the first principle calculations, the structural, electronic and optical properties of the monolayer graphene-like MoX2 sheet are calculated. Our results show that the chalcogenide atoms in the stability and the lattice parameters of the MoX2 sheet have a key role, although it is known that the electronic properties are more dependent on the metal […]
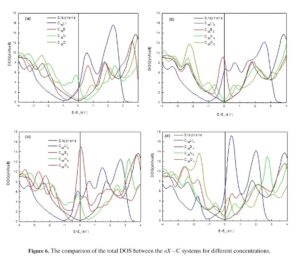
AbstractIn the context of characterizing nitrogen-poor carbonitrides for different applications, identification of an unusual onset of spin polarization of N(p) states has been shown. A full saturation up to 3 μB is demonstrated in extended two-dimensional carbon networks of NC6 and NC12 hexagonal structures refined based on density functional theory calculations. From establishing the energy–volume equations […]
AbstractNevirapine is an anti-human immunodeficiency virus (HIV) agent that belongs to the class of the non-nucleoside inhibitors of the HIV-1 virus reverse transcriptase. Spectral characteristics of nevirapine have been probed into by methods of Fourier transform infrared (FTIR), FT-Raman, UV-visible, and quantum chemistry. The UV spectrum was measured in methanol. In order to gain some […]
AbstractNew sets of molecules containing tri-phenyl-amine (TPA) core and thiophene unit with amide and imine functional groups are designed, synthesized, characterized, and compared. These are solution processable small molecules with high mobility. The newly designed molecules have better solubility due to the C=N (imine) and CONH2 (amide) moiety as compared to the established molecules with […]
AbstractAll electron density functional theory (DFT) calculations have been carried out for calcium-doped porphyrin-incorporated(5, 5) carbon nanotube (Ca-PICNT) to investigate the formation energies, electronic properties of this system, and its application in hydrogen storage. It is found that the incorporation of porphyrin ring in carbon nanotube led to a decreased value of the highest occupied […]
AbstractGround state geometry, energetics, and bonding of pure Lin ( n = 2 – 9 ) and impureLinSn ( n = 1 – 8 ) small clusters are investigated using the density functional theory. Introducing a single Sn impurity significantly changes the geometry of the host clusters for n > 5. Although the Sn atom is not trapped inside the cluster, it has the greatest coordination number among other atoms in […]
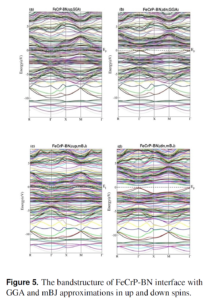
Based on the Density Functional Theory (DFT) and Generalized Gradient Approximation (GGA), the structural, electronic, and band alignment properties of the interface of FeCrP film with graphene-like BN (g-BN) were studied. These properties have been investigated at three different distances between FeCrP film and g-BN. In all three mentioned distances, the ground state point and […]