Computational study plays an important role to discover the potential of the bio-inspired nano scale molecular devices. Density Functional Theory (DFT) is one of the popular methods to calculate the properties of the molecules which can not be possible with ab initio process, preferably for transition metals. This method is important for electronic structure calculation […]
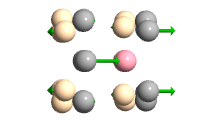
In this study the hardness SWCT was calculated with B3LYP,HF method and 3-21G,6-31G,6-311G basis set .Then it was investigated with the best method(B3LYP) and basis set(6-31G) to study the adsorption effects CO2 on the hardness of SWCNT with gap HOMO-LUMO in two shape: Horizontal, Vertical and Top-Center-Bridge and We also provide the effects of CO2 […]
In this study, B12N12 Nano ring has been selected because it consist of four 6-side rings and polar bonds B-N which in comparison with non-polar bonds C-C, is more suitable for the study of the absorption of other compounds. So reactivity and stability of Benzene alone and in the presence B12N12 nano ring field checked. To […]
Single-walled carbon nanotubes (SWCNTs) have a great deal of attention due to their unique properties. These properties of SWCNTs can be used in various devices such as nanosensors. SWCNTs nanosensors have fast response time and high sensitivity to special gas molecules which is very favorable for important applications. Recently, gas adsorption over outer surface of […]
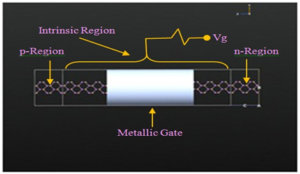
Emerging trend in semiconductor nanotechnology motivates to design various crystalline nanotubes. The structural and electronic transport properties of single walled zigzag Gallium Arsenide nanotubes have been investigated using Density Functional Theory (DFT) and Non-Equilibrium Green’s Function (NEGF) based First Principle formalisms. Structural stability and enhanced electronic transmission property of Gallium Arsenide nanotubes (NT’s) have been […]
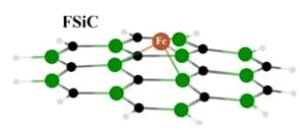
Drug delivery insights were provided by performing density functional theory (DFT) calculations to investigate the adsorption of a non-steroidal anti-inflammatory drugs; ibuprofen (IBU), by an iron-doped silicon carbide (FSiC) graphene monolayer. In this regard, the single models of IBU, SiC, and FSiC were optimized to obtain their stabilized geometries and features, in which a remarkable […]
In this research, geometrical structures of armchair single walled boron nitride nanotube (SWBNNT) and armchair single walled aluminum nitride nanotube (SWAlNNT) were optimized by Density Functional Theory (DFT) in the gas phase, both having the same length of 5 angstrom and n=9, m=9. B3LYP/6-31G* level of theory have been used to determine and compare electronic […]
Density functional theory (DFT) approach was employed to investigate relaxation processes of each of pyrimidine nucleobases (NBs); cytosine (C), thymine (T) and uracil (U), at the Cubane Cluster Surface (CCS). The main idea was about providing a material for recognition of NBs, in which a nanostructure form of cubane (CCS) was first generated by optimization […]
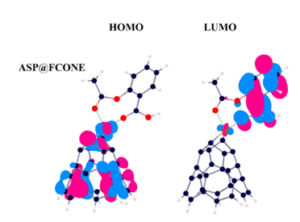
By the importance of customizing appropriate carriers for the specific drugs to approach a successful drug delivery process, the drug delivery of aspirin (ASP) was assessed by the assistance of an iron-enhanced nanocone (FCONE), using density functional theory (DFT) calculations. ASP, CONE, and FCONE models were optimized to be prepared for involving in bimolecular interactions […]
The investigation of the thermoelectric characterization of magnetic material doped Silicon Carbide (SiC) nanotube (NT) is a challenging aspect for the researchers. Thanks to the Density Functional Theory (DFT) based formalisms help to provide accurate electronic characterization for the nanoscale 2-D models. The investigation of the Device Density of States (DDOS) of the magnetically doped […]