Results and discussion
The realistic possible structure of sulphide clusters of NiS and FeS is designed in six different types. The hexagonal structure resembles bees hive like structure. The planar rhombus structure has rhombus structure which is separated by square. The cube structure has a cube-like arrangement. Planar ring structure has five transition metal atoms and five sulphur atoms which form a closed ring-like loop. In the linear ladder arrangement, all the atoms form a ladder-like structure. In the bipyramidal structure, the cube is projected with the pyramid at two faces. These are the different types of nanoclusters which are studied in the present work. The energy, dipole moment, point symmetry, HOMO–LUMO gap, ionization potential (IP), electron affinity (EA), binding energy (BE), embedding energy (EE) and vibration analysis of different sulphide clusters are analysed and reported.
Structures of (NiS)n
The possible geometrically optimized structures of (NiS)
n
are shown in the Fig.
1
. The hexagonal ring and planar rhombus have the energy of −5,718.76 and −5,718.44 Hartrees, respectively, with the dipole moment of 1.118 and 3.266 Debye, respectively. Due to closed structure of hexagonal ring the dipole moment is comparatively lower than planar rhombus. The cube structure has the energy of −7,623.87 Hartrees, cube structure has low value of 0.300 Debye which is due to the ordered structure. The planar ring, linear ladder and bipyramidal cube have the energy in the order of −9,530 Hartrees. In these structures with (NiS)
5
, the dipole moment is large for linear ladder structure whereas, the other two are almost symmetrical and have the low value of dipole moment. The obtained energy and dipole moment of geometrically optimized clusters are tabulated in Table
1
.
Structures of NiS nanoclusters Energy and dipole moment of geometrically optimized NiS nanoclusters Size Model Energy (Hartrees) Dipole moment (Debye) Symmetry 3 Hexagonal ring −5,718.76 1.1189 CS 3 Planar rhombus −5,718.44 3.2665 CS 4 Cube −7,623.87 0.3002 C1 5 Planar ring −9,531.34 0.2135 CS 5 Linear ladder −9,531.09 3.7378 CS 5 Bipyramidal cube −9,530.06 1.4671 C1Fig. 1
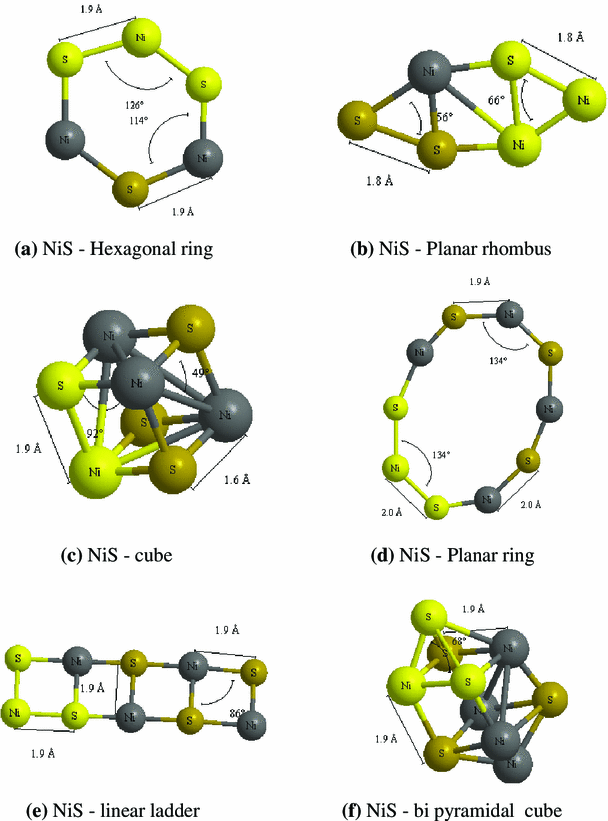
Table 1
The HOMO–LUMO gap gives the electronic properties of NiS nanoclusters [
22
,
23
]. Usually a low value of gap implies that the electron can easily move from the occupied level to the virtual level. Among all the different structures of NiS clusters, a low value is seen for linear ladder which is more suitable for electronic applications. In contrast, a high value of gap is observed for planar rhombus. In the planar rhombus Ni and S atoms are arranged symmetrically that gives rise to the high value of HOMO–LUMO gap due to overlapping of unfilled shells in
d
and
p
orbitals of Ni and S atoms. The values of HOMO–LUMO gap of NiS nanoclusters are tabulated in the Table
2
.
Energy gap and density of states of NiS nanoclusters S. No. Cluster type Energy gap(eV) NiS 1 Hexagonal ring 1.04 2 Planar rhombus 1.79 3 Cube 1.5 4 Planar ring 1.22 5 Linear ladder 0.82 6 Bipyramidal cube 1.53Table 2
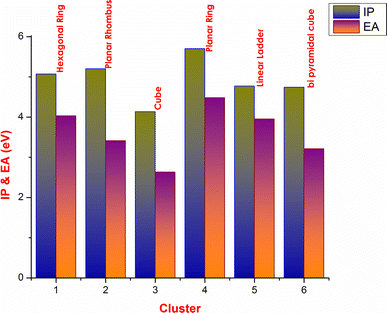
The high value of IP implies that it is difficult to remove the electron from the cluster which will be more chemically inert [
24
]. Among all the structures planar ring has the high value of IP due to the closed arrangement of atoms in the ring. EA refers the amount of energy change when the electron is added to the cluster [
25
,
26
]; a low value indicates less chemically reactive nature of the cluster. Low value of EA seen for cube structure is 2.63 eV. The IP and EA values of different clusters are shown in Fig.
2
.
Variation of IP and EA with cluster type for NiS clustersFig. 2
Structures of (FeS)n
The structures of (FeS)
n
are shown in the Fig.
3
. The calculated energies and dipole moment of various FeS nanoclusters are shown in Table
3
. The hexagonal ring and planar rhombus structure has energy of −4,984 Hartrees which has three Fe atoms and three sulphur in its clusters. In these structures a moderate dipole moment of 1.581 Debye is observed for hexagonal ring, whereas a high dipole moment of 4.165 Debye is seen for planar rhombus. In the cases of planar ring, linear ladder and bipyramidal cubes, all have the energy in and around −8,308 Hartrees with different dipole moments due to the positions of atoms: planar ring has a low dipole moment of 0.971 Debye, bipyramidal cube structure has a moderate value of 1.448 Debye and linear ladder has a high value of 5.111 Debye.
Structures of FeS nanoclusters Energy and dipole moment of geometrically optimized FeS nanoclusters Size Model Energy (Hartrees) DM (Debye) Symmetry 3 Hexagonal ring −4,984.93 1.5816 CS 3 Planar rhombus −4,984.62 4.1655 CS 4 Cube −6,645.53 0.8781 C1 5 Planar ring −8,308.3 0.9713 CS 5 Linear ladder −8,308.02 5.1115 CS 5 Bipyramidal cube −8,307.09 1.4485 C1Fig. 3
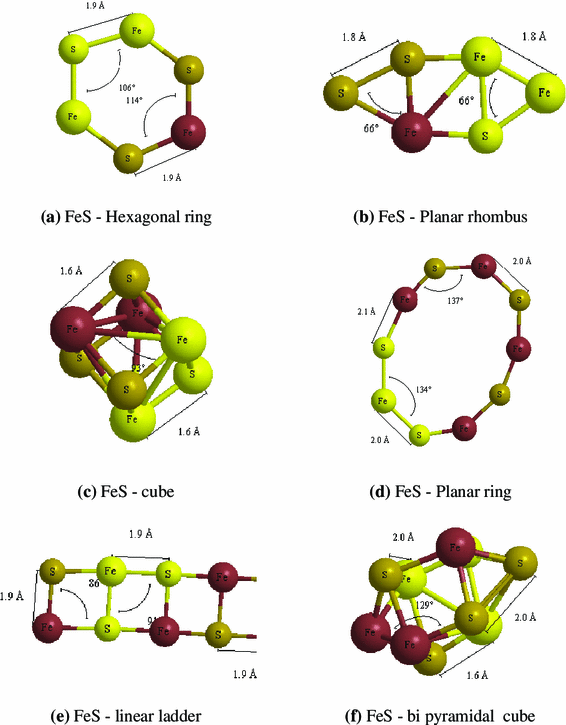
Table 3
The high value of HOMO–LUMO gap is noticed for planar rhombus; the electron requires more energy to move from HOMO to LUMO level. The high value of gap in planar rhombus structures arise due to metal–metal bonding of Fe atoms which has unoccupied
d
shells. In the case of FeS clusters, all the clusters have a low value of gap which seems to have more metallic-like property. The HOMO–LUMO gap of FeS clusters are tabulated in the Table
4
.
Energy gap and density of states of FeS nanoclusters S. No. Cluster type Energy gap(eV) FeS 1 Hexagonal ring 1.66 2 Planar rhombus 2 3 Cube 1.35 4 Planar ring 1.62 5 Linear ladder 1.61 6 Bipyramidal cube 1.33Table 4
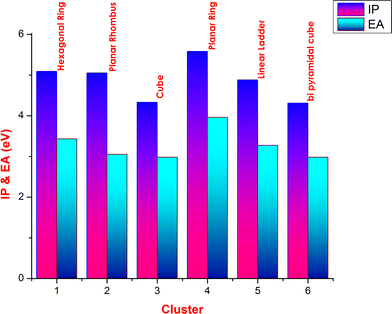
The high value of IP around 5.58 eV is noticed for planar ring due to the closed loop. Comparing with the IP value of NiS structures, the FeS clusters have low values of IP. A low value of EA is observed for cube and bipyramidal cube structures of FeS. The IP and EA variations for different clusters are shown in the Fig.
4
.
Variation of IP and EA with cluster type for FeS clustersFig. 4
Binding energies and embedding energies of (NiS)n and (FeS)n clusters
Binding energy is also one of the important parameter to analyse the structural stability of the nanoclusters [ 27 ]. The BE per atom of the nanoclusters can be calculated from the following Eq. ( 1 ). where E [TM] is the energy of the transition metal, E [S] is the energy of sulphur, E [TMS] is the energy of transition metal sulphide and n is the number of transition metal and sulphur atom in the cluster. The energy of individual atoms (TM and S) is calculated separately in the same basis set and the resultant energy values are taken into account to calculate E [TM] and E [S]. BE plays a vital role in determining the stability of the nanoclusters.
Among all the clusters of NiS, BE for cube and bipyramidal cube was high which can be said to be more stable than that of other clusters; low BE is observed for hexagonal ring and are tabulated in Table
5
. Interestingly, in the case of FeS clusters also the cube and bipyramidal cube have the high BE. The BE values of FeS nanoclusters are tabulated in Table
6
. The high value of BE for cube and bipyramidal cube ascends due to the similar kind of geometrical symmetry. Comparing the BE of all the clusters of NiS and FeS, high value is seen for FeS clusters. In NiS and FeS clusters, cube and bipyramidal cube have the high value of BE. The reason may be due to the perfect symmetry of structure between the cube and bipyramidal cube.
Calculated binding energies of NiS nanoclusters Size Model BE (eV) 3 Hexagonal ring 0.94 3 Planar rhombus 2.39 4 Cube 4.84 5 Planar ring 0.76 5 Linear ladder 1.45 5 Bipyramidal cube 4.25 Calculated binding energies of FeS nanoclusters Size Model BE (eV) 3 Hexagonal ring 1.59 3 Planar rhombus 2.99 4 Cube 5.13 5 Planar ring 1.37 5 Linear ladder 2.14 5 Bipyramidal cube 4.66Table 5
Table 6
The other parameter which can be explored is the EE; it refers to the gain in energy to embed a foreign atom during the growth process in the cluster [ 28 ]. The EE of the cluster is given by relation as given in the Eq. 2 , where TM represents transition metals such as Ni, Fe, S for sulphur and n represents the number of transition metal atoms and sulphur atom present in the clusters. For the calculation of EE, the individual energy of TM and S atoms is calculated in the same basis set and is incorporated to find E (TM) and E (S).
The variation in the EE with the cluster size of NiS is shown in the Table
7
. For NiS clusters, the bipyramidal cube structure of NiS cluster has a high value of EE which is difficult to add a foreign atom in this cluster. In FeS cluster, the EE is maximum for bipyramidal cube structure of FeS. The EE of FeS is tabulated in the Table
8
. Among all the clusters, the EE of bipyramidal cube has the high value.
Calculated embedding energies of NiS nanoclusters Size Model EE (eV) 3 Hexagonal ring 5.65 3 Planar rhombus 14.36 4 Cube 38.77 5 Planar ring 7.67 5 Linear ladder 14.50 5 Bipyramidal cube 42.50 Calculated embedding energies of FeS nanoclusters Size Model EE (eV) 3 Hexagonal ring 9.55 3 Planar rhombus 17.98 4 Cube 41.11 5 Planar ring 13.76 5 Linear ladder 21.49 5 Bipyramidal cube 46.63Table 7
Table 8
Vibrational analysis of (NiS)n and (FeS)n clusters for n = 3–5
The vibrational analysis provides the insight for the stability of the nanoclusters [
29
,
30
]. A particular cluster with a real positive frequency is said to be more stable. More IR intensity in the positive frequency leads to an increase in cluster stability. Tables
9
and
10
show the vibrational spectrum and mode assignment of NiS nanoclusters. Hexagonal ring has the significant vibrational frequency at 491.34 and 688.54 cm
−1
with the intensity of 59.75 and 27.36 km/mole both assigned to molecular stretching. In the case of planar rhombus, high IR intensity of 31.29 and 89.59 km/mole is observed at the vibrational frequency of 346.90 and 1,108.89 cm
−1
assigned to molecular stretching and S–S stretching, respectively. Looking at the vibrational frequency of cube structure, the maximum intensity of 7.35 and 21.12 km/mole for this structure is observed at the frequency of 951.55 and 1,047.37 cm
−1
respectively mode assigned to molecular stretching. The prominent IR intensity of 26.18 and 42.05 km/mole is observed for planar ring at the frequency of 28.61 and 738.04 cm
−1
respectively assigned to molecular stretching and S–Ni–S stretching. Linear ladder has the intensity of 12.09, 15.36 and 20.27 km/mole at the frequency of 636.19, 655.15 and 991.08 cm
−1
which is mode assigned to molecular twist, molecular stretching and Ni–S–Ni stretching, respectively. Bipyramidal cube has the intensity of 20.16 and 21.32 km/mole for the frequency of 634.23 and 1,070.82 cm
−1
assigned to molecular stretching and Ni–S–Ni stretching, respectively.
Vibrational spectrums of NiS nanoclusters S. No. Cluster type NiS 1 Hexagonal ring 2 Planar rhombus 3 Cube 4 Planar ring 5 Linear ladder 6 Bipyramidal cube Vibrational mode assignment of NiS nanoclusters S. No. Cluster size Model Frequency (cm−1) Intensity (km/mole) Mode assignment 1 3 Hexagonal ring 437.89 23.63 S–Ni–S stretch 491.34 59.75 Molecular stretch 688.54 27.36 Molecular stretch 2 3 Planar rhombus 346.90 31.29 Molecular stretch 1,108.89 89.59 S–S stretch 3 4 Cube 951.55 7.35 Molecular stretch 1,047.37 21.12 Molecular stretch 4 5 Planar ring 28.61 26.18 Molecular stretch 738.04 42.05 S–Ni–S stretch 1,049.28 11.66 Ni–S stretch 5 5 Linear ladder 636.19 12.09 Molecular twist 655.15 15.36 Molecular stretch 991.08 20.27 Ni–S–Ni stretch 6 5 Bipyramidal cube 634.23 20.16 Molecular stretch 1,070.82 21.32 Ni–S–Ni stretchTable 9
Table 10
The vibrational analysis of FeS is shown in Tables
11
and
12
. In the case of hexagonal ring, prominent IR intensity of 75.95 and 46.15 km/mole is observed at the frequency of 443.12 and 504.46 cm
−1
for Fe–S–Fe stretching and Fe–S–Fe bending mode respectively. The planar rhombus structure has the vibrational frequency at 741.44 and 1,118.84 cm
−1
that has the IR intensity of 12.57 and 61.50 km/mole for molecular stretching and S–S stretching respectively. The cube structure has the IR intensity of 17.24, 11.39 and 25.64 km/mole for frequency of 985.88, 1,110.02 and 1,137.97 cm
−1
for the mode of molecular bending, molecular twisting and molecular twisting, respectively. Planar ring structure has the IR intensity of 39.33, 74.09 and 104.59 km/mole for the frequency of 570.5, 795.6 and 906.13 cm
−1
assigned to molecular stretching, S–Fe–S stretching and Fe–S–Fe stretching, respectively. In the case of linear ladder, the vibrational frequency occurs at 671.1, 714.71 and 752.79 cm
−1
with IR intensity of 38.13, 330.66 and 42.85 km/mole which is assigned to molecular stretching, molecular bending and Fe–S stretching, respectively. The bipyramidal cube has the vibrational frequency of 440.66, 656.47 and 1,160.71 cm
−1
with IR intensity of molecular twist, molecular stretching and Fe–S stretching.
Vibrational spectrums of FeS nanoclusters S. No. Cluster type FeS 1 Hexagonal ring 2 Planar rhombus 3 Cube 4 Planar ring 5 Linear ladder 6 Bipyramidal cube Vibrational mode assignment of FeS nanoclusters S. No. Cluster size Model Frequency (cm−1) Intensity (km/mole) Mode assignment 1 3 Hexagonal ring 443.12 75.95 Fe–S–Fe stretch 504.46 46.15 Fe–S–Fe bend 737.89 15.62 Fe–S bend 2 3 Planar rhombus 741.44 12.57 Molecular stretch 1,118.84 61.50 S–S stretch 3 4 Cube 985.88 17.24 Molecular bend 1,110.02 11.39 Molecular twist 1,137.97 25.64 Molecular twist 4 5 Planar ring 570.50 39.33 Molecular stretch 795.60 74.09 S–Fe–S stretch 906.13 104.59 Fe–S–Fe stretch 5 5 Linear ladder 671.1 38.13 Molecular stretch 714.71 330.66 Molecular bend 752.79 42.85 Fe–S stretch 6 5 Bipyramidal cube 440.66 17.48 Molecular twist 656.47 17.58 Molecular stretch 1,160.71 34.72 Fe–S stretchTable 11
Table 12