Graphical abstract

An efficient and sustainable method was developed for the electrocatalytic synthesis of nano-sized 4 H -pyran derivatives via a green, one-pot, three-component condensations of cyclic-1,3-diketones [including 1H-indene-1,3(2H)-dione, cyclohexane-1,3-dione, cyclopentane-1,3-dione, pyrimidine-2,4,6(1H,3H,5H)-trione], malononitrile or ethyl cyanoacetate, and isatins in an undivided electrochemically cell applying potassium bromide as the electrolyte in the alcoholic media. This electrosynthesis was introduced as a reliable and cost-effective approach for the high-yielding synthesis (76–92%) of the target compounds. The synthesized 4 H -pyran and chromene nanoparticles could open an effective way for production of nano-sized drugs.
Nowadays the invention of fast, environmentally friendly, and inexpensive synthetic protocols have become a main part of research in organic and medicinal chemistry which is due to the facile preparation of compounds with biologically activities [ 1 , 2 ]. The electrocatalytic multicomponent reaction has been known as an important approach to address this issue is, in which three or more starting materials are combined in an electrochemical cell with the appropriate electrolyte and working electrodes to generate the target products [ 3 , 4 ]. Regarding to the recent growing interest of researchers in applying electrochemistry in organic compound synthesis, the electrosynthesis has been known as one of the most viable protocols in modern organic chemistry which offers a novel and promising versatile synthetic method for organochemists [ 5 ]. The electrochemical procedure permits the valuable synthesis of organic compounds for the large-scale processes due to its catalytic nature and also use of available, inexpensive, and environmentally-friendly chemical compounds such as electrolyte [ 6 , 7 ]. The electrosynthesis of heterocyclic compounds at mild conditions such as ambient temperature and pressure make it highlight in organic synthesis. Additionally, the catalytic amount of an electrogenerated base can efficiently induce the catalytic chain transformation of organic reactants into target components. All of these aspects find in good agreement with the important principles in green chemistry [ 8 ].
4 H -Pyran derivatives have been as a valuable class of heterocyclic compounds, which are extensively distributed in nature [ 9 ]. The fused pyran ring skeleton is considered as a famous heterocyclic compound and also as a significant framework of many natural products. In recent years, pyran and fused pyran derivatives have received the most attention owing to their association with the various types of biological and pharmaceutical characteristics [ 10 ]. The Pyran derivatives reveal interesting medicinal properties, including antimicrobial [ 11 ], antifungal [ 12 ], antitumor [ 13 ], anticoagulant, and anti-anaphylactic activities [ 14 ]. Moreover, these heterocyclic compounds exhibit potentially positive effects in the treatment of the neurodegenerative diseases such as Parkinson, Huntington, and Alzheimer, as well as AIDS-associated dementia [ 15 ]. These heterocyclic compounds are also applied in preparation of cosmetics, pigments and photoactive materials [ 16 ]. The drug structures with high surface-volume ratio display substantial improvement of solubility which cause stronger therapeutic effect. So, nano- or micro-sized drugs due to their high surface-volume ratio cause increasing of the drug adsorption and improving the curative characteristic. Accordingly, the development of several new methods to synthesize the nano-sized drugs is an significant challenge for both chemist and pharmacist. Several methods, including micronization, modification of polymorphic configuration, expansion of oil-based solutions, smart application of co-solvents, application of stabilizing agents, microemulsions, and creation of solid dispersions have been offered for the synthesis of nano-sized drug compounds.
Following our previous efforts to develop the new procedures for synthesis of the biologically active heterocyclic compounds from inexpensive and available chemical reagents [
17
,
18
–
19
], herein, a new, green, and convenient protocol was described for the synthesis of pyran derivatives based on the electrocatalytic multicomponent reaction. In this study, electrochemically induced catalytic three-component condensation of cyclic-1,3-diketones, malononitrile/ethyl cyanoacetate with isatins in KBr-contained ethanol were applied to synthesize the nanoparticles of pyran derivatives (Scheme
1
).
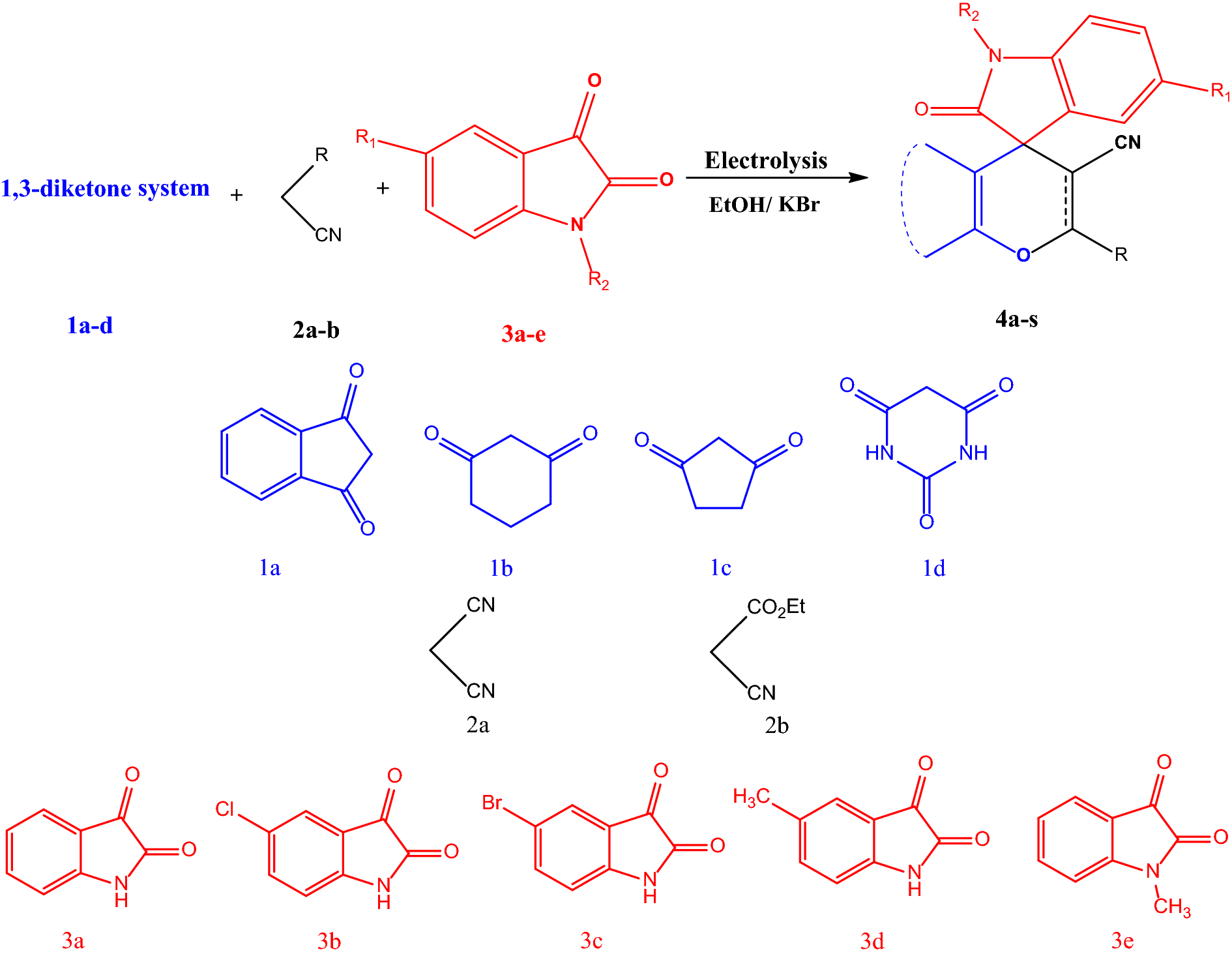
The electrosynthesis of the pyran-derivatives nanoparticles using cyclic-1,3-diketones (
1a–d
), malononitrile (
2a
) or ethyl cyanoacetate (
2b
), and isatines (
3a–e
)Scheme 1
All consumed chemical materials and solvents were acquired from the Merck and Sigma-Aldrich companies. The melting points of products were evaluated by a melting point apparatus of IA 9100. Controlled-current coulometry and preparative electrolysis were performed via a SAMA potentiostat/galvanostat (Isfahan-Iran). The platinum cathode (5 cm 2 ), graphite anode (5 cm 2 ), as well as iron cathode, and magnesium anode were used as working electrodes. The infrared spectra (IR) were procured using the Bruker Vector 22 FTIR in KBr disks. The scanning electron microscopy images were acquired using the Hitachi S-4160 machine, which was made in Japan. The 1 H NMR spectrums were achieved in DMSO- d 6 using the Bruker-Arance AQS 500 MHz and the 13 C NMR spectrums were recorded in DMSO- d 6 employing a Bruker-Avance spectrometer 125 MHz. In addition, the mass spectrums were determined by Agilent Technology (HP) mass spectrometer operating at an ionization potential of 70 eV. Resulting products refined using thin layer chromatography (TLC) on silica gel PolyGram SILG-UV 254 plates for more purification.
A mixture of cyclic 1,3-diketones ( 1a–d ) (1 mmol), malononitrile ( 2a ) or ethyl cyanoacetate ( 2b ) (1 mmol), isatins ( 3a–e ) (1 mmol), and potassium bromide (0.5 mmol) in alcohol (20 ml) was electrolyzed in an undivided cell supplied with a magnetic stirrer, iron cathode (5 cm 2 ), magnesium anode (5 cm 2 ) at the temperature of 40 °C and using catalytic quantity at the constant current density (20 mA cm −2 , I = 100 mA) and passing the 0.1 F mol −1 of electricity through the system (time: 50 min). The reaction progression was controlled by TLC analysis (ethyl acetate/ n -hexane 1/3). After the reaction was complete, the reaction mixture become cold to the ambient temperature, then it was condensed under the reduced pressure to one-fifth of initial volume.
The remaining solid was separated through filtration, next it was washed with cold ether and dried at room temperature. The resulting nano-sized products were determined through several techniques.
White solid, (yield 92%), m.p 247–249 °C, IR (KBr) ( ν max /cm −1 ) 3337 (NH 2 ), 2904 (NH), 2223 (CN), 1712 (C=O), 1623 (C=O); 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 6.88–7.16 (m, 4H, Ar), 7.23 (s, 2H, NH 2 ), 7.43–7.66 (m, 4H, Ar),10.43 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 45.2–56.9–103.4–117.8–121.7–125.2–126.9–127.8–127.9–128.5–129.7–129.9–134.5–136.3–137.8–141.7–160.1–168.1–171.4–193.1.
White solid, (yield 90%), m.p 302–304 °C, IR (KBr) ( ν max /cm −1 ) 3347 (NH 2 ), 3160 (NH), 2196 (CN), 1717 (C=O), 1658 (C=O); 1 HNMR (500 MHz, DMSO-d 6 ): δ (ppm): 1.58 (m, 2H, CH 2 ), 1.96 (t, 2H, CH 2 ), 2.83 (t, 2H, CH 2 ), 6.88–7.42 (m, 4H, Ar), 7.31 (s, 2H, NH 2 ), 10.24 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 19.2–28.6–36.5–45.4–58.8–107.8–116.9–122.0–125.2–127.7–127.9–130.1–141.0–155.1–160.1–168.7–200.3
White solid, (yield 89%), m.p 303–305 °C, IR (KBr) ( ν max /cm −1 ) 3339 (NH 2 ), 3142 (NH), 2183 (CN), 1711 (C=O), 1641 (C=O); 1 HNMR (500 MHz, DMSO-d 6 ): δ (ppm): 1.87 (t, 2H, CH 2 ), 3.01 (t, 2H, CH 2 ),6.93-7.41 (m, 4H, Ar), 7.36 (s, 2H, NH 2 ), 10.44 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 28.3–32.2–46.9–58.3–108.7–117.6–122.6–125.1–127.8–128.2–130.0–142.4–159.1–160.4–169.3–203.7
White solid, (yield 92%), m.p 252–254 °C, 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 1.91 (t, 2H, CH 2 ), 2.18 (t, 2H, CH 2 ), 2.66 (t, 2H, CH 2 ), 3.14 (s, 3H, CH 3 ), 6.96–7.24 (m, 4H, Ar),7.28 (s, 2H, NH 2 ); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 20.20–26.80–27.18–36.77–46.99–57.27–108.53–112.28–117.67–122,83–123.39–128.81–134,13–149.96–159.20–166.60–177.09–195.45.
White solid, (yield 92%), m.p 247–249 °C, 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 0.79 (t, 3H, CH 3 ), 1.88 (t, 2H, CH 2 ), 2.19 (m, 2H, CH 2 ),2.64 (t, 2H, CH 2 ), 3.70 (q, 2H, CH 2 ), 6.66–7.05 (m, 4H, Ar), 7.86 (s, 2H, NH 2 ), 10.15 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 13.75–20.11–27.39–38.56–59.27–108.48–114.70–120.96–122.88–127.57–136.57–144.49–159.45–164.64–168.12–180.33–195.24.
White solid, (yield 92%), m.p 236–238 °C, 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 0.73 (t, 3H, CH 3 ), 1.88 (t, 2H, CH 2 ), 2.17 (m, 2H, CH 2 ),2.65 (t, 2H, CH 2 ), 3.11 (s, 3H, CH 3 ), 3.66 (q, 2H, CH 2 ), 6.83–7.17 (m, 4H, Ar), 7.89 (s, 2H, NH 2 ); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 13.95–20.08–26.55–27.36–37.46–59.18–107.22–114.61–121.76–122.69–127.87–135.62–145.71–159.61–164.85–167.97–178.70–195.32.
Yellow solid, (yield 92%), m.p 282-285 °C; 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 6.78–7.17 (m, 4H, Ar), 7.34 (s, 2H, NH 2 ), 10.46 (s, 1H, NH), 11.06(s, 1H, NH),12.20 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 46/96–58.26–86.77–109.36–117.32–122.29–123.95–128.61–133.88–142.57–149.84–153.84–158.79–162.11–178.07.
White solid, (yield 92%), m.p 229–232 °C; 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 0.78 (s, 3H, CH 3 ), 3.71 (q, 2H, CH 2 ), 6.67–7.08 (m, 4H, Ar), 7.97 (s, 2H, NH 2 ), 10.03 (s, 1H, NH),12.18 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 13.30–22.58–44.19–58.78–89.19–108.38–121.09–123.05–127.64–135.32–149.61–152.25–158.41–161.88–167.68–179.57.
White solid, (yield 92%), m.p 308–311 °C; 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 3.14 (s,3H,CH 3 ), 6.90–7.29 (m, 4H, Ar), 7.40 (s, 2H, NH 2 ), 11.08 (s, 1H, NH),12.26 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 26.72–87.05–108.05–110.71–122.82–123.78–125.93–129.03–133.10-138.04–144.04–149.75–154.01–158.95–161.77–176.60.
White solid, (yield 92%), m.p 236–238 °C; 1 HNMR (500 MHz, DMSO-d6): δ (ppm): 0.72 (s, 3H, CH 3 ), 3.02 (s, 3H, CH 3 ), 3.60 (q, 2H, CH 2 ), 6.85–7.19 (m, 4H, Ar), 7.97 (s, 2H, NH 2 ), 10.03 (s, 1H, NH),12.18 (s, 1H, NH); 13 C-NMR (125 MHz, DMSO-d6): δ (ppm): 13.49–56.22–61.56–76.46–89.79–108.68–120.64–123.27–127.64–135.53–144.19–145.84–149.54–152.85–158.29–161.80–167.75–179.71.
In order to continue our previous attempts for electrosynthesis of heterocyclic compounds [ 20 ], in this work, a new strategy based on the electrocatalytic three-component transformation of cyclic-1,3-diketones ( 1a–d ), malononitrile ( 2a ) or ethyl cyanoacetate ( 2b ), and isatins ( 3a-e ) was reported for the synthesis of nano-sized pyran derivatives. The procedure was performed using constant-current electrolysis in an undivided cell with KBr as the supporting electrolyte. First, to optimize the reaction conditions, the three-component reactants, including cyclic-1,3-diketones (2 H -indene-1,3-dione) ( 1a ), malononitrile ( 2a ), and isatin ( 3a) were followed in alcoholic media inside of an undivided cell as a pattern reaction.
The reaction was examined in different conditions such as different temperatures, several solvents (ethanol, methanol, and n-propanol) and various applied current. The platinum cathode along with the graphite anode in ethanol could upgrade the higher yields synthesis of pyran derivatives at shorter time with the current density of 20 mA cm
−2
(
I
= 100 mA, electrode surface = 5 cm
2
). Ethanol was preferred as an alcoholic solvent at the temperature of 40 °C, instead of propanol and methanol. Electrolysis of cyclic-1,3-diketones (
1a–d
), malononitrile (
2a
) or ethyl cyanoacetate (
2b
), and isatins (
3a–e
) in ethanol at the constant current produced the nano-sized pyran derivatives (
4a–s
) (Table
1
). In the next step, magnesium and iron were chosen as the anode and the cathode electrodes respectively in the different alcoholic electrolytes (Table
2
). Then, the obtained products were defined using scanning electron microscopy (SEM) which verified the nano-sized
4H
-pyran derivative synthesis in the form of tunable nanoparticles (Fig.
1
). It is suggested that the application of magnesium as the anode electrode and iron as the cathode electrode might be causing the production of the nano-sized pyran derivatives. The optimized current density and temperature had no significant changes compared to the previous works, but the synthesis duration of
4a
decreased from 65 min in previous woks to 50 min in this work (Table
2
). In this research, to expand the area of the reaction, a comprehensive study was carried out on a wide range of pyran derivatives and almost all of the pyran derivatives were synthesized at the nano-sized scale. The electrolysis of the starting materials with magnesium as the anode and iron as the cathode in ethanol at the constant current (20 mA cm
−2
), were led to nano-sized pyran derivatives (
4a–s
) with 76
–
92% yields (Tables
1
,
3
).
Electrosynthesis of nano-sized
4H
-pyran derivatives (
4a–s
) in ethanol at the constant current (20 mA cm
−2
) at 50 min The analogy of the current and the solvent effect on the reaction of 1,3-indanedione (
1a
), malononitrile (
2a
), and isatin (
3a
) resulting in the pyran-derivative nanoparticles (
4a
) Entry Solvent (20 ml) Temperature (°C) Current density (mA cm−2) Time (min) Electricity passed (Fmol−1) Yield (%) 1 MeOH r.t 30 6 50 0.1 54 2 MeOH 35 30 6 25 0.1 62 3 n-PrOH r.t 30 6 50 0.1 59 4 n-PrOH 35 30 6 25 0.1 70 5 EtOH r.t 30 6 50 0.1 75 6 EtOH 35 30 6 25 0.1 72 7 EtOH 35 100 20 50 0.1 84 8 EtOH 40 100 20 50 0.1 92 9 EtOH 45 200 40 80 0.1 71 10 EtOH 50 250 50 100 0.1 56 11 EtOH 50 250 50 30 0.1 50 The SEM image of 2-amino-2′,5-dioxo-5
H
-spiro(indeno(1,2-b)pyran-4,3′-indoline)-3-carbonitrile nanoparticles The results obtained from the synthesis of nano-sized pyran derivatives (
4a–s
) Entry cyclic-1,3-diketones Yield (%) Time (min) Electricity passed (Fmol−1) MP (°C) Lit. MP (°C) [ 1a 92 50 0.1 247–249 245 1b 90 50 0.1 302–304 > 300 1c 89 50 0.1 303–305 > 300 1a 81 50 0.1 226–228 – 1b 84 50 0.1 291–292 288–290 1c 83 50 0.1 302–304 > 300 1a 79 50 0.1 223–225 220 1b 76 50 0.1 299–301 298–300 1c 80 50 0.1 299–301 > 300 1a 89 50 0.1 237–239 – 1b 85 50 0.1 293–295 292–294 1c 83 50 0.1 305–306 > 300 1b 89 50 0.1 252–254 – 1b 86 50 0.1 252–254 253–256 1b 85 50 0.1 236–238 – 1d 88 50 0.1 282–285 – 1d 84 50 0.1 229–232 – 1d 86 50 0.1 308–311 – 1d 83 50 0.1 236–238 –Table 1
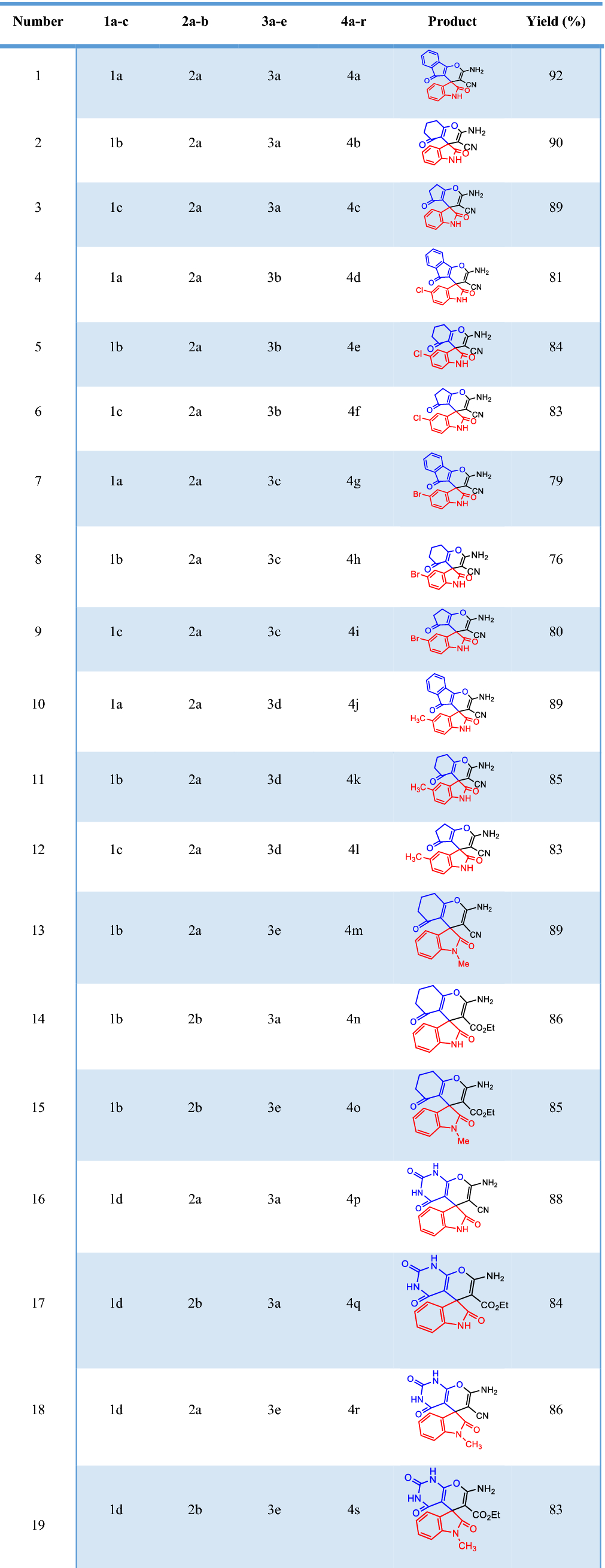
Table 2
Fig. 1

Table 3
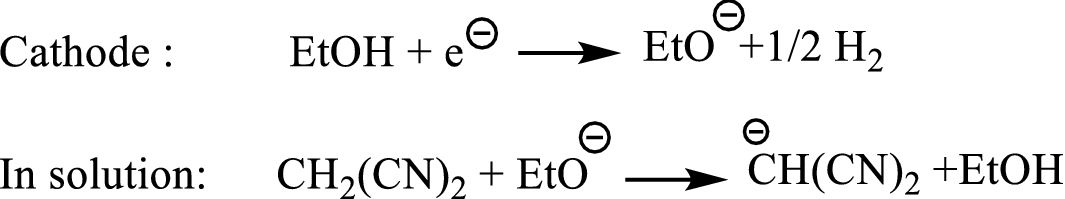
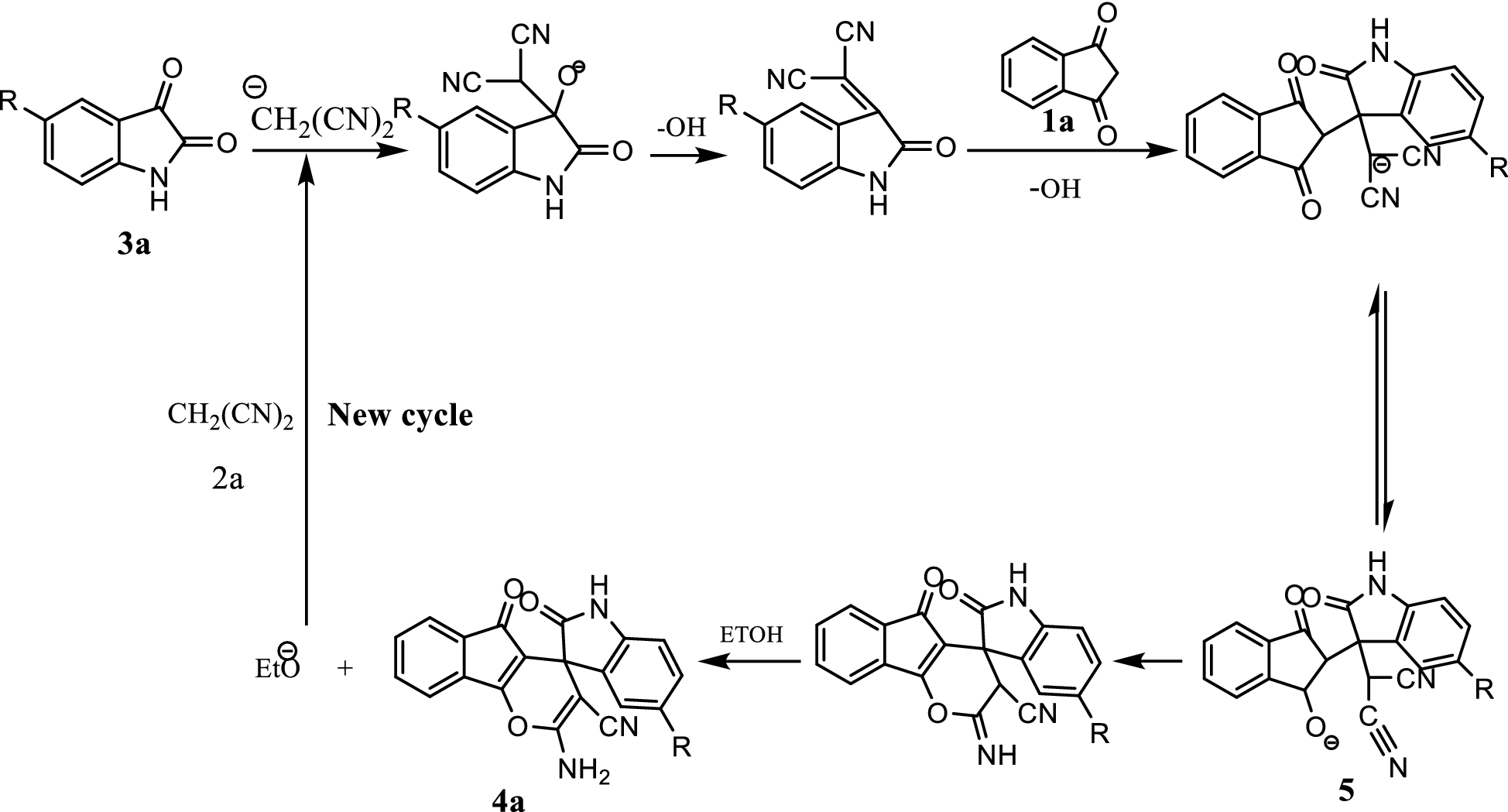
At the first step, deprotonation of ethanol occurred in cathode which led to the formation of an ethoxide anion (Scheme
2
). The next reaction occurred between the ethoxide anion and malononitrile, producing the malononitrile anion. Then, Knoevenagel condensation of the malononitrile anion with isatins (
3a
) took places through the removal of a hydroxide anion and formation of 2-(2-oxoindolin-3-ylidene)malononitrile (Scheme
3
). The following hydroxide-promoted Michael addition of cyclic-1,3-diketones such as 1,3-Indandione (
1a
) resulted in the intermediate (
5
) which was followed by intramolecular cyclization and production of the corresponding 4
H
-pyran derivatives
4
through the regeneration of the ethoxide anion as the cyclic starting of the reaction with malononitrile.
Formation of ethoxide anion at the cathode The mechanism of the electrocatalytic three-component transformation of 1,3-indanedione (
1a
), malononitrile (
2a
), and isatin (
3a
)Scheme 2
Scheme 3
The introduced nanoparticles of pyran derivatives were synthesized using a simple and effective electrosynthesis approach under the mild, rapid, and selective conditions. Besides the excellent yields of target compounds, the electrocatalytic procedure offers a reasonable combination of conventional three-component reactions with convenient ecological advantages and facile electrochemical methods. This method could be evaluated as a noteworthy alternative approach due to the mentioned advantages. The produced nanoparticles of 4H -pyran derivatives could be very effective especially in pharmacy and medicine.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.