Graphical abstract

A series of first-, second-, and third-generation dendronic triazolo-pyridazinones were synthesized in good yields via the Cu I -catalyzed azide–alkyne cycloaddition reactions of 4,6-diphenyl-2-(prop-2-yn-1-yl)pyridazin-3(2 H )-one, possessing a terminal alkyne functional group with aromatic mono- and diazides with long alkyl and chiral glycol side-chain substituents. The chemical structures of the new compounds were characterized using different spectroscopic methods. The morphology of the dendrons was examined using the scanning electron microscope (SEM) analysis, which revealed the formation of highly ordered nanofiber and nanorod aggregations, directed by π-stacking interactions and van der Waals forces.
Molecular self-assembly (MSA) has been ubiquitous in supramolecular chemistry, material sciences, and nanoscience [ 1 , 2 – 3 ]. It describes the phenomenon in which smaller components come spontaneously and precisely together with the aid of noncovalent forces, such as hydrogen bonding [ 4 ], charge-transfer interactions in electron-rich π-donor and electron-deficient π-acceptor systems [ 5 ], aromatic π-stacking through molecular orbital overlapping [ 6 ], van der Waals forces, [ 7 ] or metal-ion interactions [ 8 ] to construct well-ordered aggregations and thereby create order out of disorder. As a consequence, the entropic barrier of the organized transition state of the MSA aggregations tends to decrease in comparison with the chaotic monomeric species. The microscopic analysis outcome of the MSA is usually visualized as aggregations of nanofibers [ 9 ], nanorods, [ 10 ] nanotubes [ 11 ], or nanospheres [ 12 ]. MSA has been commonly observed in nature, for instance, in the formation of the various intriguing biomolecules, such as the DNA nucleobases, proteins, viruses, and lipid bilayers [ 13 , 14 , 15 – 16 ]. A growing interest has been dedicated over the past decades to two-dimensional (2D) self-assembly of supramolecular architectures at solid surfaces due to its impact for developing new nanomaterials [ 17 ]. The MSA of materials at solid–liquid surfaces is counterbalanced by many factors, such as the material concentration in the dispersion medium [ 18 ], change of pH, or metal complexation [ 19 , 20 ], leading probably to alteration of the mode of assembly. Recently, MSA directed by hydrophobic–hydrophilic interactions has been given a widespread research interest due to its directionality and selectivity [ 21 , 22 , 23 – 24 ]. Inspired by our recent work on the self-assembly of dendrimers [ 19 , 20 , 25 ], we designed and prepared a series of first-, second-, and third-generation dendrons to investigate the roles of aromatic π-stacking interactions and the weak van der Waals forces in controlling the surface morphology of the self-organized materials.
The microanalytical data were carried out on a Vario El-Mentar instrument, Microanalytical Center, Cairo University, Egypt. The 1 H-NMR and 13 C-NMR spectra were measured on a JEOL 300 MHz in DMSO- d 6 , Microanalytical Center, Cairo University, Egypt. The chemical shifts ( δ ) were expressed in ppm relative to the standard TMS as an internal reference. Mass spectra were measured on a 70 eV EI Ms-QP 1000 EX equipment (Shimadzu, Japan), Microanalytical Center, Cairo University, Egypt. The UPLC separations were performed using a Waters Alliance system (mixtures and gradient mixtures of acetonitrile/water, flow = 0.6 ml/min) equipped with 100 × 2.1 mm AQUITY HSST3 column (1.8 µm phenyl-hexyl material) and consisted of a Waters Separations Module 2695, a Waters Diode Array Detector 996, and a Waters Mass Detector ZQ 2000. Conditions are specified when describing the corresponding compounds. The signals have been detected using UV between 200 and 400 nm (MaxPlot). The SEM measurements were performed using a Quanta FEG 250 instrument, National Research Centre, Egypt.
A three-necked flask was charged with alkyne 7 (1 equiv.), aryl azide 5 , 8 , 11 , 13 , 15 , or 17 (1 equiv.), sodium ascorbate (0.3 equiv.), TBTA (0.15 equiv.), and a solvent mixture of H 2 O/ t BuOH/CH 2 Cl 2 (1/2/8, 60 ml). The flask was evacuated and flushed with argon repeatedly (three cycles). Thereafter, CuSO 4 ·5H 2 O was added (0.15 equiv.) and the reaction mixture was stirred for 2 days at rt in the dark. After consumption of alkyne 7 as indicated by TLC monitoring, the mixture was diluted with CH 2 Cl 2 and transferred into a separating funnel for extraction. The organic phase was separated and washed with an aqueous solution of ethylenediaminetetraacetic acid (EDTA) disodium salt (EDTA-Na 2 ) (3×). The aqueous phase was extracted with CH 2 Cl 2 (3×) and washed with a saturated aqueous solution of NaCl (60 ml volume). The organic phase was dried over anhydrous MgSO 4 and filtered. The solvent was evaporated under vacuo and the target compound was obtained after purification with column chromatography.
In a two-necked flask equipped with a condenser, pyrogallol 1 (1.0 g, 8 mmol, 1 equiv.), 1-bromohexadecane (7.33 g, 24 mmol, 3 equiv.), 18-crown-6 (0.31 g, 1.2 mmol, 0.15 equiv.), tetrabutylammonium iodide (0.44 g, 1.2 mmol, 0.15 equiv.), and K 2 CO 3 (8.29 g, 60 mmol, 7.5 equiv.) were dissolved in 60 ml acetonitrile. The mixture was degassed at rt by evacuating under stirring, flushed with argon (3×), and refluxed overnight. After consumption of pyrogallol as indicated by TLC monitoring, the solution was transferred into a separating funnel and diluted with ethyl acetate (EA) for extraction. The organic phase was washed with a saturated aqueous solution of NaHCO 3 (3×), water (3×), and brine. The organic phase was dried over anhydrous MgSO 4 , filtered, and the solvent was evaporated under vacuo to give the target compound after purification with column chromatography [petroleum ether (PE): EA 2:1]. Yield: 99%, 1 H-NMR (300 MHz, CDCl 3 ): δ (ppm) = 0.90 (t, 3 J = 6.49 Hz, 9H, C H 3 ), 1.23–1.49 (m, 78H, C H 2 ), 1.74–1.87 (m, 6H, C H 2 ), 3.95–4.0 (m, 6H, C H 2 ), 6.57 (d, 3 J = 8.34 Hz, 2H, Ar H ), and 6.92 (t, 3 J = 8.33 Hz, 1H, Ar H ). 13 C-NMR (75 MHz, CDCl 3 ): δ (ppm) = 14.13 (C H 3 ), 22.73 (C H 2 ), 26.15 (C H 2 ), 26.19 (C H 2 ), 28.22 (C H 2 ), 28.81 (C H 2 ), 29.42 (C H 2 ), 29.48 (C H 2 ), 29.59 (C H 2 ), 29.70 (C H 2 ), 29.72 (C H 2 ), 29.76 (C H 2 ), 30.39 (C H 2 ), 31.97 (C H 2 ), 32.88 (C H 2 ), 33.90 (C H 2 ), 69.07 (OC H 2 ), 73.36 (OC H 2 ), 106.75 ( C Ar ), 123.10 ( C Ar ), 138.44 (CO), and 153.43 (CO). MS (EI, T = 37–50 °C): m / z = 800.31 (Calcd. 800.39 for [M+H + ]). Elemental analysis, calcd. (%) for C 54 H 102 O 3 : C 81.13, H 12.86; found C 81.17, H 12.79.
1,2,3-Tris(hexadecyloxy)benzene 2 (0.5 g, 0.6 mmol, 1 equiv.) was dissolved in 15 ml CH 2 Cl 2 , and an aqueous solution of sodium nitrite was added (0.08 g, 1.25 mmol, 2 equiv.). The suspension was cooled to 0 °C, and 2 ml nitric acid 65% was added dropwise over the course 20 min. The mixture was stirred at rt for 3 h and 100 ml of PE was added. The organic layer was washed with a saturated solution of NaHCO 3 , brine, dried over anhydrous MgSO 4 , and filtered over a small pad of silica gel using CH 2 Cl 2 as an eluent; the solvent was evaporated under vacuo and the residue was dried overnight using a vacuum pump. Yield: 71%, 1 H-NMR (300 MHz, CDCl 3 ): δ (ppm) = 0.90 (t, 3 J = 6.50 Hz, 9H, C H 3 ), 1.45–1.51 (m, 78H, C H 2 ), 1.71–1.90 (m, 6H, C H 2 ), 4.03–4.10 (m, 6H, OC H 2 ), and 7.49 (s, 2H, Ar H ). 13 C-NMR (75 MHz, CDCl 3 ): δ (ppm) = 14.11 (C H 3 ), 22.71 (C H 2 ), 26.04 (C H 2 ), 28.21 (C H 2 ), 28.80 (C H 2 ), 29.12 (C H 2 ), 29.40 (C H 2 ), 29.48 (C H 2 ), 29.54 (C H 2 ), 29.58 (C H 2 ), 29.65 (C H 2 ), 30.33 (C H 2 ), 31.96 (C H 2 ), 32.87 (C H 2 ), 33.86 (C H 2 ), 69.40 (OC H 2 ), 73.76 (OC H 2 ), 102.06 ( C Ar ), 143.13 (NO 2 C Ar ), 143.77 (CO), and 152.69 (CO). MS (EI, T = 37–50 °C): m / z = 845.43 (Calcd. 845.39 for [M+H + ]). Elemental analysis, calcd. (%) for C 54 H 101 NO 5 : C 76.81, H 12.06, N 1.66; found C 76.85, H 12.11, N 1.98.
In a one-necked flask, 1,2,3-tris(hexadecyloxy)-5-nitrobenzene 3 (4.36 g, 5.17 mmol, 1 equiv.) was dissolved in 50 ml of methanol, 0.430 mg Pd/C was added, and the mixture was degassed at rt under vacuo and flushed with H 2 (3x). After stirring overnight at 60 °C under an atmosphere of H 2 , the mixture was filtered through a Celite pad and the solvent was evaporated under vacuo. Yield: 91%, 1 H-NMR (300 MHz, CDCl 3 ): δ (ppm) = 0.90 (t, 3 J = 6.49 Hz, 9H, C H 3 ), 1.2–1.4 (m, 78H, C H 2 ), 1.7–1.8 (m, 6H, C H 2 ), 3.8–3.9 (m, 6H, OC H 2 ), 4.13–4.18 (brs, 2H, NH 2 ), and 5.99 (s, 2H, Ar H ). 13 C-NMR (75 MHz, CDCl 3 ): δ (ppm) = 14.13 (C H 3 ), 22.71 (C H 2 ), 26.12 (C H 2 ), 26.21 (C H 2 ), 29.40 (C H 2 ), 29.44 (C H 2 ), 29.68 (C H 2 ), 29.74 (C H 2 ), 30.32 (C H 2 ), 31.95 (C H 2 ), 68.97 (OC H 2 ), 73.58 (OC H 2 ), 94.56 ( C Ar ), 131.19 (CO), 141.96 (NH 2 C Ar ), and 153.72 (CO). MS (EI, T = 37–50 °C): m / z = 815.48 (Calcd. 815.41 for [M+H + ]). Elemental analysis, calcd. (%) for C 54 H 103 NO 3 : C 79.64, H 12.75, N 1.72; found C 79.69, H 12.71, N 1.78.
In a round-bottomed flask, 3,4,5-tris(hexadecyloxy)aniline 4 (2.48 g, 3 mmol, 1 equiv.) was dissolved in 10 ml of acetonitrile and cooled to 0 °C in an ice bath. To the stirred mixture was added t -BuONO (0.46 g, 4.5 mmol, 1.5 equiv.) followed by dropwise addition of TMSN 3 (0.41 g, 3.6 mmol, 1.2 equiv.). The resulting solution was stirred at rt for 1 h. The reaction mixture was concentrated under vacuo , and the crude product was purified by silica gel chromatography using PE to give the product. 1 H-NMR (300 MHz, CDCl 3 ): δ (ppm) = 0.90 (t, 3 J = 6.48 Hz, 9H, C H 3 ), 1.28–1.49 (m, 78H, C H 2 ), 1.72–1.84 (m, 6H, C H 2 ), 3.90–3.98 (m, 6H, OC H 2 ), and 6.23 (s, 2H, Ar H ). 13 C-NMR (75 MHz, CDCl 3 ): δ (ppm) = 14.13 (C H 3 ), 22.72 (C H 2 ), 26.07 (C H 2 ), 26.14 (C H 2 ), 29.30 (C H 2 ), 29.40 (C H 2 ), 29.66 (C H 2 ), 29.74 (C H 2 ), 29.76 (C H 2 ), 30.30 (C H 2 ), 31.95 (C H 2 ), 69.15 (OC H 2 ), 73.55 (OC H 2 ), 97.65 ( C Ar ), 134.97 (CO), 135.46 (N 3 C Ar ), and 153.92 (CO). MS (EI, T = 37–50 °C): m / z = 841.31 (Calcd. 841.41 for [M+H + ]). Elemental analysis, calcd. (%) for C 54 H 101 N 3 O 3 : C 77.18, H 12.11, N 5.00; found C 77.25, H 12.06, N 5.04.
In a two-necked flask fitted with an efficient reflux condenser was dissolved 4,6-diphenylpyridazin-3(2 H )-one 6 (10 mmol) in DMF (20 ml) followed by addition of Na 2 CO 3 (30 mmol). The reaction mixture was heated to 50–60 °C for 30 min and then cooled to rt. Propargyl bromide 80% in toluene (20 mmol) was added. The reaction mixture was stirred at the same temperature and the reaction progress was monitored by TLC. After consumption of the starting material (about 4 h), the mixture was poured into ice water (100 ml) with stirring. The resulting solution was extracted with Et 2 O (100 ml) (3×). The combined organic extracts were washed with cold water (100 ml) (2×) and dried over anhydrous MgSO 4 . The organic solvent was evaporated under vacuo to give the title compound as a pale yellow material (94%). TLC (PE/EA 8/2) R f = 0.39. 1 H-NMR (300 MHz, DMSO- d 6 ): δ (ppm) = 3.27 (s, 1H, C≡C H ), 5.03 (s, 2H, C H 2 ), 7.47–7.52 (m, 6H, Ar H ), 7.94–8.01 (m, 4H, Ar H ), and 8.15 (s, 1H, Ar H ). 13 C-NMR (75 MHz, DMSO- d 6 ): δ (ppm) = 38.68 ( C H 2 ), 75.24 (C≡C H ), 78.46 ( C ≡C H ), 126.02 ( C Ar ), 127.32 ( C Ar ), 128.03 ( C Ar ), 128.73 ( C Ar ), 128.87 ( C Ar ), 129.34 ( C Ar ), 129.40 ( C Ar ), 133.65 ( C Ar ), 134.36 ( C Ar ), 138.63 ( C Ar ), 144.23 ( C Ar ), and 157.75 (CO). MS (EI, T = 37–50 °C): m / z = 287.34 (Calcd. 287.34 for [M+H + ]). Elemental analysis, calcd. (%) for C 19 H 14 N 2 O: C 79.70, H 4.93, N 9.78; found C 79.75, H 4.88, N 9.84.
The title compound was separated and purified by column chromatography (PE/EA, 7/3) as a white solid (89%). TLC (PE/EA, 7/3) R f = 0.48. 1 H-NMR (300 MHz, DMSO- d 6 ): δ (ppm) = 0.82 (t, 3 J = 6.55 Hz, 6H, C H 3 ), 1.19–1.32 (m, 52H, C H 2 ), 1.77–1.82 (m, 4H, C H 2 ), 3.96 (t, 3 J = 6.76 Hz, 4H, CO 2 C H 2 ), 5.58 (s, 2H, C H 2 ), 6.99–7.48 (m, 6H, Ar H ), 7.63–8.28 (m, 8H, Ar H +C5- H ), and 8.65 (s, 1H, Ar H ). 13 C-NMR (75 MHz, DMSO- d 6 ): δ (ppm) = 14.11 ( C H 3 ), 22.69 ( C H 2 ), 25.98 ( C H 2 ), 28.61 ( C H 2 ), 29.25 ( C H 2 ), 29.36 ( C H 2 ), 29.54 ( C H 2 ), 29.61 ( C H 2 ), 29.70 ( C H 2 ), 31.91 ( C H 2 ), 50.43 ( C H 2 ), 65.98 (O C H 2 ), 118.65 ( C Ar ), 119.20 ( C Ar ), 124.81 ( C Ar ), 127.52 ( C Ar ), 128.47 ( C Ar ), 128.64 ( C Ar ), 128.69 ( C Ar ), 128.84 ( C Ar ), 129.07 ( C Ar ), 130.64 ( C Ar ), 131.85 ( C Ar ), 133.17 ( C Ar ), 133.84 ( C Ar ), 135.24 ( C Ar ), 144.10 ( C Ar ), 149.32 ( C Ar ), 157.70 ( C O), and 165.21 ( C O 2 ). MS (EI, T = 37–50 °C): m / z = 965.31 (Calcd. 965.31 for [M + Na + ]). Elemental analysis, calcd. (%) for C 59 H 83 N 5 O 5 : C 75.20, H 8.88, N 7.43; found C 75.14, H 8.82, N 7.35.
The title compound was separated and purified by column chromatography (PE/EA, 7/3) as a white solid (86%). TLC (PE/EA 7/3) R f = 0.39. 1 H-NMR (300 MHz, DMSO- d 6 ): δ (ppm) = 0.85 (t, 3 J = 7.03 Hz, 9H, C H 3 ), 1.20–1.35 (m, 78H, C H 2 ), 1.80–1.85 (m, 6H, C H 2 ), 3.85–3.93 (m, 6H, OC H 2 ), 5.57 (s, 2H, C H 2 ), 7.23–7.33 (m, 5H, Ar H ), 7.49–7.56 (m, 5H, Ar H ), and 7.92–8.12 (m, 4H, Ar H +C5- H ). 13 C-NMR (75 MHz, DMSO- d 6 ): δ (ppm) = 14.12 ( C H 3 ), 22.53 ( C H 2 ), 25.94 ( C H 2 ), 27.68 ( C H 2 ), 29.28 ( C H 2 ), 29.34 ( C H 2 ), 29.56 ( C H 2 ), 29.60 ( C H 2 ), 29.73 ( C H 2 ), 31.87 ( C H 2 ), 50.72 ( C H 2 ), 70.53 (O C H 2 ), 74.12 (O C H 2 ), 102.82 ( C Ar ), 119.31 ( C Ar ), 122.59 ( C Ar ), 123.37 ( C Ar ), 127.69 ( C Ar ), 128.45 ( C Ar ), 128.67 ( C Ar ), 128.78 ( C Ar ), 128.97 ( C Ar ), 129.11 ( C Ar ), 130.01 ( C Ar ), 131.84 ( C Ar ), 131.89 ( C Ar ), 135.52 ( C Ar ), 140.27 (O C Ar ), 144.15 ( C Ar ), 148.65 (O C Ar ), 156.42 ( C Ar ), and 158.31 ( C O). MS (EI, T = 37–50 °C): m / z = 1127.73 (Calcd. 1127.73 for [M+H + ]). Elemental analysis, calcd. (%) for C 73 H 115 N 5 O 4 : C 77.82, H 10.29, N 6.22; found C 77.86, H 10.24, N 6.17.
The title compound was separated and purified by column chromatography (PE/EA, 8/2) as a beige solid (79%). TLC (PE/EA, 8/2) R f = 0.36. 1 H-NMR (300 MHz, DMSO- d 6 ): δ (ppm) = 1.27 (d, 2 J = 6.2 Hz, 3H, CHC H 3 ), 3.10 (s, 3H, OC H 3 ), 3.25–3.41 (m, 14H, OC H 2 ), 5.24–5.33 (m, 1H, C H CH 3 ), 5.57 (s, 4H, C H 2 ), 7.39–7.42 (m, 10H, Ar H ), 7.47 (d, 2 J = 2.1 Hz, 4H, Ar H ), 7.91–7.96 (m, 7H, Ar H ), 8.08 (m, 2H, C5- H ), 8.62 (s, 2H, Ar H ), and 9.03 (s, 2H, Ar H ). 13 C-NMR (75 MHz, DMSO- d 6 ): δ (ppm) = 16.20 (CHC H 3 ), 47.71 ( C H 2 ), 57.87 (O C H 3 ), 69.44 ( C HCH 3 ), 69.65 (O C H 2 ), 69.73 (O C H 2 ), 70.06 (O C H 2 ), 71.12 (O C H 2 ), 71.26 (O C H 2 ), 72.52 (O C H 2 ), 115.50 ( C Ar ), 119.57 ( C Ar ), 122.83 ( C Ar ), 126.00 ( C Ar ), 127.04 ( C Ar ), 127.98 ( C Ar ), 128.68 ( C Ar ), 128.91 ( C Ar ), 129.25 ( C Ar ), 129.38 ( C Ar ), 132.95 ( C Ar ), 133.75 ( C Ar ), 134.43 ( C Ar ), 137.61 ( C Ar ), 138.56 ( C Ar ), 143.67 ( C Ar ), 144.10 ( C Ar ), 158.21 ( C O), and 163.32 ( C O 2 ). MS (EI, T = 37–50 °C): m / z = 982.11 (Calcd. 982.07 for [M+H + ]). Elemental analysis, calcd. (%) for C 55 H 52 N 10 O 8 : C 67.33, H 5.34, N 14.28; found C 67.39, H 5.28, N 14.34.
In a one-necked round-bottomed flask, (4 mmol, 1 equiv.) of 3,5-diaminobenzoic acid was dissolved in 18% HCl under heating with a heat gun and the mixture was cooled to 0 °C in an ice bath. Sodium nitrite (8.4 mmol, 2.1 equiv.) was added in small portions. The mixture was stirred for 15 min at 0 °C, then NaN 3 (8.8 mmol, 2.2 equiv.) was added in small portions and the stirring was continued for 30 min, while the temperature was maintained at 0 °C. The mixture was poured into 400 ml ice water, filtered, and the solid was washed with water. The title compound was obtained as a colorless solid (86%) and was found to be sufficiently pure for characterization without further purification. 1 H-NMR (300 MHz, D 2 O): δ (ppm) = 7.01 (s, 1H, Ar H ), 7.33 (s, 2H, Ar H ), and 13.43 (s, 1H, CO 2 H ). 13 C-NMR (75 MHz, D 2 O): δ (ppm) = 113.98 ( C Ar ), 116.00 ( C Ar ), 133.92 ( C Ar ), 141.46 ( C Ar ), and 165.73 ( C O 2 ).
The title compound was separated and purified by column chromatography (PE/EA, 7/3) as a beige solid (75%). TLC (PE/EA, 7/3) R f = 0.41. 1 H-NMR (300 MHz, DMSO- d 6 ): δ (ppm) = 5.59 (s, 4H, C H 2 ), 7.45–7.50 (m, 11H, Ar H ), 7.93–7.99 (m, 10H, Ar H ), 8.14 (m, 2H, C5- H ), 8.65 (s, 2H, Ar H ), 9.06 (s, 2H, Ar H ), and 13.21 (s, 1H, CO 2 H ). 13 C-NMR (75 MHz, DMSO- d 6 ): δ (ppm) = 47.69 ( C H 2 ), 114.25 ( C Ar ), 116.14 ( C Ar ), 122.83 ( C Ar ), 124.64 ( C Ar ), 126.10 ( C Ar ), 127.21 ( C Ar ), 128.04 ( C Ar ), 128.37 ( C Ar ), 128.77 ( C Ar ), 128.98 ( C Ar ), 129.32 ( C Ar ), 129.41 ( C Ar ), 133.83 ( C Ar ), 134.52 ( C Ar ), 138.67 ( C Ar ), 143.73 ( C Ar ), 144.17 ( C Ar ), 158.26 ( C O), and 164.89 ( C O 2 ). MS (EI, T = 37–50 °C): m / z = 777.35 (Calcd. 777.81 for [M+H + ]). Elemental analysis, calcd. (%) for C 45 H 32 N 10 O 4 : C 69.58, H 4.15, N 18.03; found C 69.53, H 4.21, N 17.95.
The title compound was separated and purified by column chromatography (PE/EA, 6/4) as a beige solid (82%). TLC (PE/EA, 6/4) R f = 0.55 1 H-NMR (300 MHz, DMSO- d 6 ): δ (ppm) = 1.01 (q, J = 7.5 Hz, 3H, SiC H CH 3 ), 1.09 (d, 2 J = 1.8 Hz, 18H, SiCHC H 3 ), 1.33 (d, 2 J = 6.0 Hz, 3H, CHC H 3 ), 3.20 (s, 3H, OC H 3 ), 3.28 (s, 3H, OC H 3 ), 3.36–3.60 (m, 22H, OC H 2 ), 3.91 (t, 3 J = 4.9 Hz, 2H, OC H 2 ), 4.19 (t, 3 J = 4.6 Hz, 2H, OC H 2 ), 5.22–5.29 (m, 1H, C H CH 3 ), 5.63 (s, 2H, C H 2 ), 7.08 (d, 2 J = 2.1 Hz, 2H, Ar H ), 7.29 (d, 2 J = 2.5 Hz, 2H, Ar H ), 7.43–7.45 (m, 5H, Ar H ), 7.96–7.99 (m, 3H, Ar H ), 8.16 (s, 1H, C5- H ), 8.54, 8.56 (ss, 2H, Ar H ), 9.02 (s, 2H, Ar H ), and 9.50 (s, 1H, Ar H ). 13 C-NMR (75 MHz, CDCl 3 ): δ (ppm) = 11.06 (Si C H), 16.27 (CH C H 3 ), 18.45 (SiCH C H 3 ), 47.79 ( C H 2 ), 57.91 (O C H 3 ), 57.99 (O C H 3 ), 68.60 (O C H 2 ), 69.48 (O C H 2 ), 69.61 (O C H 2 ), 69.70 (O C H 2 ), 69.78 (O C H 2 ), 70.00 (O C H 2 ), 70.08 (O C H 2 ), 71.17 (O C H 2 ), 71.26 (O C H 2 ), 71.35 (O C H 2 ), 72.56 (O C H 2 ), 90.28 ( C ≡C), 106.08 ( C ≡C), 106.50 ( C Ar ), 115.12 ( C Ar ), 117.25 ( C Ar ), 119.68 ( C Ar ), 120.59 ( C Ar ), 120.70 ( C Ar ), 122.33 ( C Ar ), 122.75 ( C Ar ), 126.10 ( C Ar ), 127.17 ( C Ar ), 127.70 ( C Ar ), 128.01 ( C Ar ), 128.76 ( C Ar ), 128.98 ( C Ar ), 129.41 ( C Ar ), 133.11 ( C Ar ), 133.85 ( C Ar ), 134.52 ( C Ar ), 137.66 ( C Ar ), 138.65 ( C Ar ), 143.51 ( C Ar ), 143.85 ( C Ar ), 144.19 ( C Ar ), 147.56 ( C Ar ), 151.15 ( C Ar ), 158.27 ( C O), and 165.44 ( C O 2 ). MS (EI, T = 37–50 °C): m / z = 1141.21 (Calcd. 1141.41 for [M+H + ]). Elemental analysis, calcd. (%) for C 61 H 77 N 9 O 11 Si: C 64.25, H 6.81, N 11.05; found C 64.28, H 6.87, N 10.98.
The title compound was separated and purified by column chromatography (CH 2 Cl 2 /CH 3 OH, 100/2) as a yellow solid (69%), TLC (CH 2 Cl 2 /CH 3 OH, 100/2) R f = 0.36. 1 H-NMR (300 MHz, CDCl 3 ): δ (ppm) = 1.22 (d, 2 J = 5.57 Hz, 24H, CHC H 3 ), 3.09 (s, 24H, OC H 3 ), 3.14–3.23 (m, 112H, C H 2 ), 5.21 (s, 8H, CO 2 C H ), 5.58 (s, 2H, C H 2 ), 7.19–7.27 (m, 8H, Ar H ), 7.45–7.58 (m, 6H, Ar H ), 7.95 (s, 2H, Ar H ), 8.13 (s, 1H, C5- H ), 8.20–8.32 (m, 11H, Ar H ), and 8.67–8.95 (m, 8H, Ar H ). 13 C-NMR (75 MHz, CDCl 3 ): δ (ppm) = 16.15 (CH C H 3 ), 50.59 ( C H 2 ), 58.25 (O C H 3 ), 65.73 ( C H 2 ), 69.98 ( C H 2 ), 70.08 ( C H 2 ), 70.42 ( C H 2 ), 70.89 ( C H 2 ), 72.18 ( C H 2 ), 74.35 ( C H 2 ), 117.06 ( C Ar ), 117.43 ( C Ar ), 120.12 ( C Ar ), 126.11 ( C Ar ), 121.68 ( C Ar ), 123.45 ( C Ar ), 125.34 ( C Ar ), 127.42 ( C Ar ), 128.07 ( C Ar ), 128.57 ( C Ar ), 128.71 ( C Ar ), 128.89 ( C Ar ), 129.15 ( C Ar ), 129.54 ( C Ar ), 130.21 ( C Ar ), 132.26 ( C Ar ), 133.80 ( C Ar ), 134.71 ( C Ar ), 135.18 ( C Ar ), 136.02 ( C Ar ), 137.02 ( C Ar ), 139.87 ( C Ar ), 141.61 ( C Ar ), 142.03 ( C Ar ), 145.42 ( C Ar ), 152.75 ( C Ar ), 157.82 ( C O), and 164.51 ( C O 2 ). UPLC R t = 5.82, 67% peak area. HRMS (ESI) m/z = 3254.466 (Calcd. 3254.482 for [M+H + ]). Elemental analysis, calcd. (%) for C 158 H 206 N 26 O 49 : C 58.33, H 6.38, N 11.19; found C 58.27, H 6.45, N 11.12.
We have utilized the Cu
I
-catalyzed azide–alkyne cycloaddition reaction to prepare the dendronic materials under investigation in this work. To investigate the role of van der Waals forces in controlling the MSA of the dendronic triazolo-pyridazinones, long alkyl side chains were introduced into the dendronic framework through azide
5
, which was prepared in a stepwise reaction as shown in Scheme
1
.
Synthetic pathway of the azidobenzene
5
and pyridazin-3(2
H
)-one
7Scheme 1
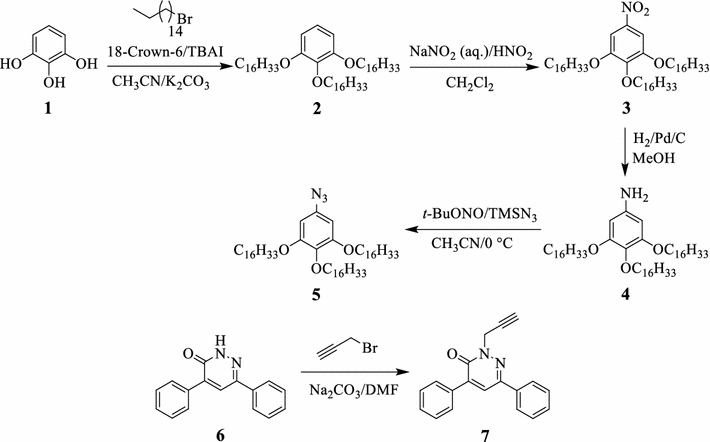
Pyrogallol 1 reacted with 1-bromohexadecane in acetonitrile to afford the 1,2,3-tris(hexadecyloxy)benzene 2 , which upon nitration gave the 1,2,3-tris(hexadecyloxy)-5-nitrobenzene 3 . Thereafter, the 1,2,3-trisubstituted nitrobenzene 3 underwent hydrogenation over Pd/C catalyst in methanol to yield the corresponding 3,4,5-tris(hexadecyloxy)aniline 4 , which was subsequently reacted with tert -butyl nitrite and trimethylsilyl azide to furnish the corresponding 5-azido-1,2,3-tris(hexadecyloxy)benzene 5 . The chemical structure of azidobenzene 5 was determined using the various spectroscopic characterization techniques. The 1 H-NMR of 5 showed the expected signals of the aliphatic chains at δ 0.90–3.98 ppm and the two symmetric aromatic signals at δ 6.23 ppm. The electron impact (EI) mass spectrum of 5 showed the expected protonated molecular ion at m / z 841.31 as [M+H + ]. On the other hand, pyridazin-3(2 H )-one 7 , possessing two phenyl moieties and a free terminal alkyne group, was prepared in excellent yield (94%) via the basic treatment of 4,6-diphenylpyridazin-3(2 H )-one 6 [ 27 ] with propargyl bromide in DMF. The 1 H-NMR spectrum of 7 showed two singlet signals at δ 3.27 ppm and δ 5.03, corresponding to the acetylenic C H and C H 2 protons, respectively. The EI mass spectrum showed the anticipated protonated molecular ion [M+H + ] at m / z 287.34. The dihexadecyl-5-azidoisophthalate 8 with two long-chain alkyl side chains was prepared according to the reported synthetic procedure [ 28 ].
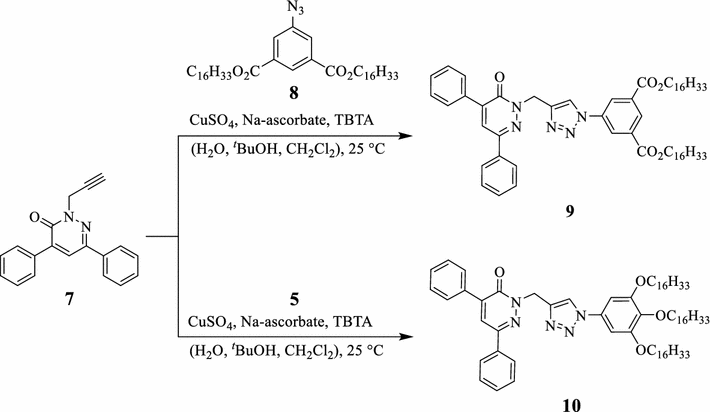
The Cu
I
-catalyzed 1,3-dipolar cycloaddition reactions of aromatic azides
5
and
8
with the terminal alkyne functional group of the pyridazinone
7
afforded the first-generation G1 dendrons
9
and
10
, possessing two and three long alkyl chains and two phenyl substituents loaded on the pyridazinone ring (Scheme
2
). The 1,2,3-triazole rings function as junction units between the phenyl-substituted pyridazinones and the di- or tri-alkyl-substituted phenyl rings. The
1
H-NMR spectrum of
9
, for example, showed the signals corresponding to the alkyl chains at
δ
0.82–3.96 ppm, in addition to the singlet C
H
2
protons at
δ
5.58 ppm. The C
H
signal of the junction 1,2,3-triazole appeared downfield shifted, overlapping with the aromatic protons, at
δ
7.63–8.28 ppm. The
13
C-NMR spectrum of
9
showed the signal of the carbonyl group of the pyridazinone ring at
δ
157.70 ppm, while the signal of the carbonyl group of the ester group appeared at
δ
165.21 ppm. The EI mass spectrum showed the molecular ion peak at
m
/
z
965.31 as [M+Na
+
]. On the other side, click chemistry was employed to prepare the second-generation G2 dendrons
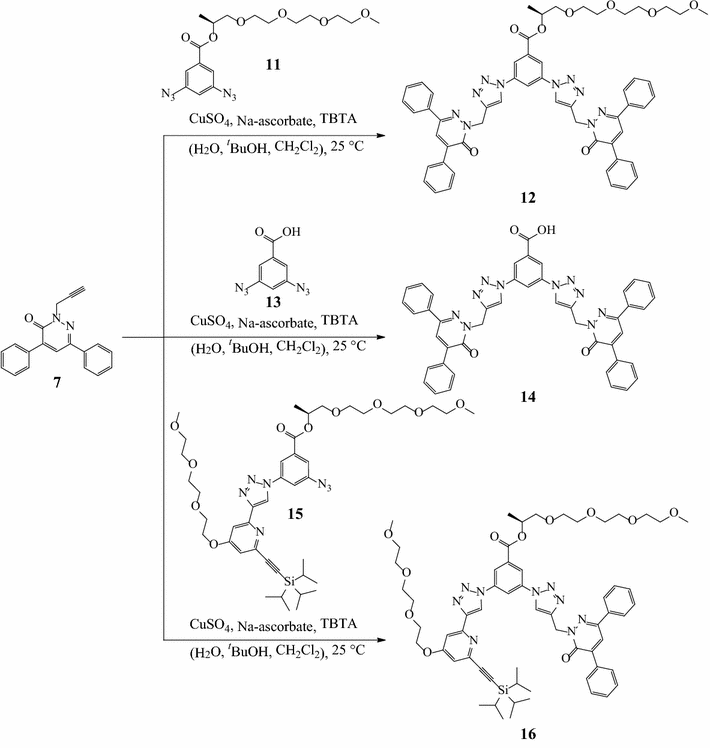
12
,
14
, and
16
. The click reactions of the terminal alkyne moiety of the pyridazinone
7
with the diazides
11
[
29
] and
13
afforded dendrons
12
and
14
, thus introducing four phenyl rings to the dendronic structural framework to enhance the intermolecular π-stacking interactions as demonstrated in Scheme
3
. The non-symmetric azide
15
was prepared according to the literature [
29
]. As similar, the click reaction of the pyridazinone
7
with azide
15
afforded the second-generation G2 dendron
16
.
Click synthesis of the first-generation dendrons
9
and
10 Click synthesis of the second-generation dendrons
12
,
14
, and
16Scheme 2
Scheme 3
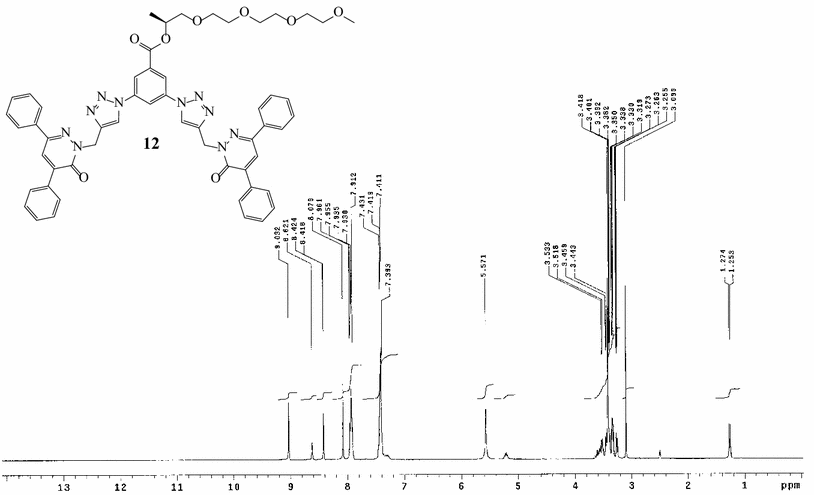
The
1
H-NMR spectrum of dendron
12
, for instance, showed the expected signals of the chiral glycol side chain at
δ
1.27–5.33 ppm and the singlet C
H
2
at
δ
5.57 ppm. The singlet C
H
signal of the junction 1,2,3-triazole appeared downfield shifted at
δ
8.08 ppm (Fig.
1
). The
13
C-NMR spectrum of dendron
12
showed the signal of the carbonyl group of the pyridazinone ring and the carbonyl group of the glycol ester functionalities at
δ
158.21 and 163.32 ppm, respectively. The EI mass spectrum confirmed the molecular weight of dendron
12
via the appearance of the characteristic peak of the protonated molecular ion at
m
/
z
982.11 as [M+H
+
]. The third-generation G3 dendron
17
[
30
] with a terminal azide group underwent click reaction with the terminal alkyne group of pyridazinone
7
to give dendron
18
(Scheme
4
). The
1
H-NMR spectrum of
18
showed the signals of the chiral glycol chains at
δ
1.22–5.21 ppm and the singlet C
H
2
signal at
δ
5.58 ppm. The singlet signal of the junction 1,2,3-triazole ring appeared at
δ
8.13 ppm. The
13
C-NMR spectrum of dendron
18
showed the signal of the carbonyl group of the pyridazinone ring and the carbonyl group of the glycol ester group at
δ
157.82 and 164.51 ppm, respectively. The high-resolution electrospray ionization (ESI) mass spectrum of dendron
18
showed the anticipated protonated molecular ion peak at
m/z
3254.466 as [M+H
+
]. The surface morphology of dendrons
9
,
10
,
12
,
14
, and
18
was investigated using the scanning electron microscope (SEM) analysis to investigate the roles of π-stacking and van der Waals forces in controlling the self-assembly of the dendronic materials. The dendrons
9
and
10
were self-assembled to form well-ordered and uniform nanofibrous aggregations as shown in Fig.
2
. The self-assembly was most probably directed cooperatively by intermolecular π-stacking of the phenyl rings and the weak van der Waals forces of the long alkyl substituents.
1
H-NMR spectrum of dendron
12
(300 MHz, DMSO-
d
6
) Click synthesis of the third-generation dendron
18 Scanning electron microscope examinations (SEM) of the morphology of dendrons
a 9
(10 and 5 µm, average width 458.6–992.7 nm),
b 10
(10 and 5 µm, average width 286.8–449.7 nm),
c 12
(4 and 1 µm, average dimensions 146.1 × 176.7 nm), and
d 14
(5 and 2 µm, average width 87.17–151.9 nm)Fig. 1
Scheme 4
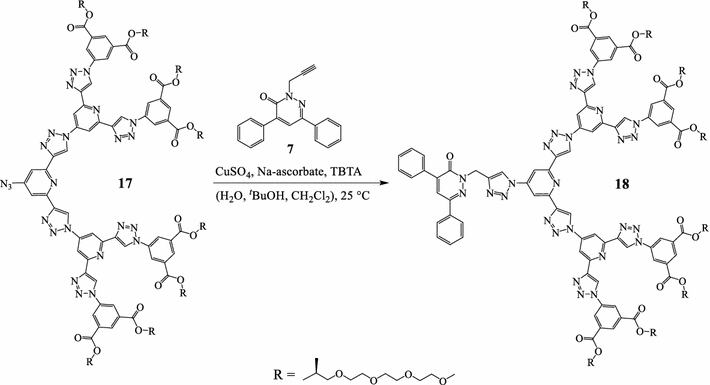
Fig. 2

The second-generation dendrons 12 and 14 incorporated four benzene rings in their structures to reinforce the intermolecular π-stacking interactions. However, they exhibited considerably different morphological features. The SEM examination of the dendron 14 revealed the formation of long extended and entwined nanofibers via π-stacking interactions, occurring between the phenyl substituents, as the main driving force of the intermolecular self-assembly, while dendron 12 formed interesting nanorod aggregations with a well-defined rectangle structure. As similar to dendron 14 , the mode of assembly displayed by the dendron 12 was controlled by the intermolecular π-stacking interactions and the chirality introduced through the glycol chain, which was expected additionally to direct the MSA to form the rectangle nanorods. Unfortunately, the third-generation dendron 18 did not show any interesting morphological features as the role of the π-stacking interactions of the two phenyl rings might not be effectively relative to the structure of the dendron. The literature is rich with many examples which demonstrate the morphological dependence on the secondary noncovalent interactions of the self-assembled materials [ 31 , 32 , 33 , 34 , 35 , 36 , 37 – 38 ]. A recent example was reported by Yam and co-workers on the MSA of the platinum(II) 2,6- bis (benzimidazol-2-yl)pyridine double complex salts via metal–metal and π–π stacking interactions to form 1D infinite chains in water [ 39 ]. Another example was reported by Palanisamy et al. in which three weak noncovalent interactions, including van der Waals forces between the alkyl side chains, hydrogen bonding, and π-stacking, participated cooperatively in the MSA of aromatic biscarbamate architectures into xerogel fibers [ 40 ].
The Cu I -catalyzed azide–alkyne cycloaddition reactions have been utilized to furnish the first-, second-, and third-generation dendrons via reacting 4,6-diphenyl-2-(prop-2-yn-1-yl)pyridazin-3(2 H )-one 7 , which possess free terminal alkyne group with aromatic mono- and diazides, possessing alkyl and glycol chain substituents. We have demonstrated the unique self-assembling ability of a series of alkyl- and aromatic-substituted dendrons into nanofiber and nanorod materials. The roles displayed by aromatic π-stacking and van der Waals interactions in controlling the morphology of the dendrons were investigated by the SEM analysis. The formation of extended fibers in case of dendrons 9 and 10 was most likely directed by cooperative aggregations resulted from the π-stacking interactions of the benzene groups and the van der Waals interactions between the long alkyl side-chain substituents. On the other side, the main driving force for the MSA of dendrons 12 and 14 into highly ordered nanorods with well-defined rectangle morphology or nanofibers was expected to be the π-stacking interactions of the aromatic substituents. The 4,6-diphenylpyridazin-3(2 H )-one 6 has been shown to be a versatile precursor for the synthesis of both nanofibrous and nanorod materials through the introduction of the long alkyl chains via the click chemistry approach and the aromatic substituents loaded on the molecule.
The authors gratefully acknowledge the financial support of the sector of research projects at the National Research Centre, Cairo, Egypt (Project No. AR110101).
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.