Results and discussion
Fibrillar semiconductive P3HT single crystals
Use of the self-seeding method led to completely uniform population in the semiconductive fibrillar P3HT single crystals. Figure
1
a, b illustrates atomic force microscopy (AFM) height image of P3HT
48800
single crystals grown from xylene at
T
s
= 40 °C,
T
c
= 30 °C and TEM image accompanied by the corresponding selected area electron diffraction (SAED) pattern. The dimensions of these single crystals in the
a
[thickness of single crystal or hexyl side chains or (100) direction],
b
[length of single crystal or π–π stacking or (020) direction], and
c
(width of single crystal or longitude of P3HT main backbones) axes were 437.50 nm, 64.55 µm, and 116.87 nm, respectively. In the SAED patterns of Fig.
1
b, two pairs of (100) prisms in the
a
-axis or the hexyl side chains direction and two pairs of (020) prisms in the
b
-axis or the π–π stacking direction demonstrated a flat-on orientation for the P3HT chains, in which the main backbones and the hexyl side chains were perpendicular and parallel to the substrate, respectively. The schemes of Fig.
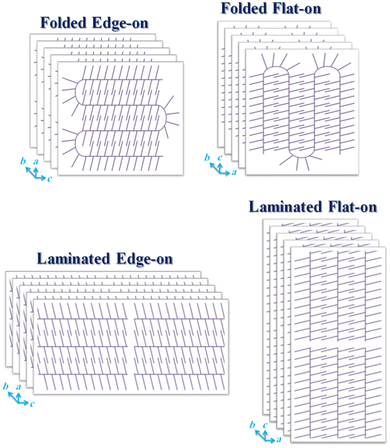
2
depict the folded flat-on (top, right) and laminated flat-on (bottom, right) P3HT chains for the high molecular weight (48,000 g/mol) and low molecular weight (21,000 and 7150 g/mol) P3HTs, respectively. Likewise, AFM height image of P3HT
21000
single crystals grown from toluene at
T
s
= 30 °C,
T
c
= 20 °C and TEM image with the corresponding SAED pattern in the inset are represented in Fig.
1
c, d. The similarity from the perspectives of thickness, width, and length of fibrillar single crystals was detected in this growth system as well. The dimensions of these single crystals in the
a
,
b
, and
c
directions were 95.00 nm, 36.75 µm, and 100.55 nm, respectively. In the SAED pattern of the fibrillar P3HT single crystals depicted in Fig.
1
d, (020) prisms in the π–π stacking direction and (002) in the longitude of P3HT backbone were detected. This pattern was indicative of an edge-on orientation, in which the P3HT main backbones and the hexyl side chains were parallel with and perpendicular to the substrate, respectively. The schemes of Fig.
2
represent the folded edge-on (top, left) and laminated edge-on (bottom, left) P3HT chains for the high molecular weight (48,000 g/mol) and low molecular weight (21,000 and 7150 g/mol) P3HTs, respectively.
AFM height image (
a
) and TEM image accompanied by the corresponding SAED pattern in the
inset
(
b
) for P3HT
48800
single crystals grown from xylene at
T
s
= 40 °C,
T
c
= 30 °C; AFM height image (
c
) and TEM image accompanied by the corresponding SAED pattern in the
inset
(
d
) for P3HT
21000
single crystals grown from toluene at
T
s
= 30 °C,
T
c
= 20 °C Scheme of backbone folding in high molecular weight P3HTs in edge-on-oriented (
top
,
left
) and flat-on-oriented (
top
,
right
) single crystals as well as backbone lamination in low molecular weight P3HTs in edge-on-oriented (
bottom
,
left
) and flat-on-oriented (
bottom
,
right
) single crystalsFig. 1

Fig. 2
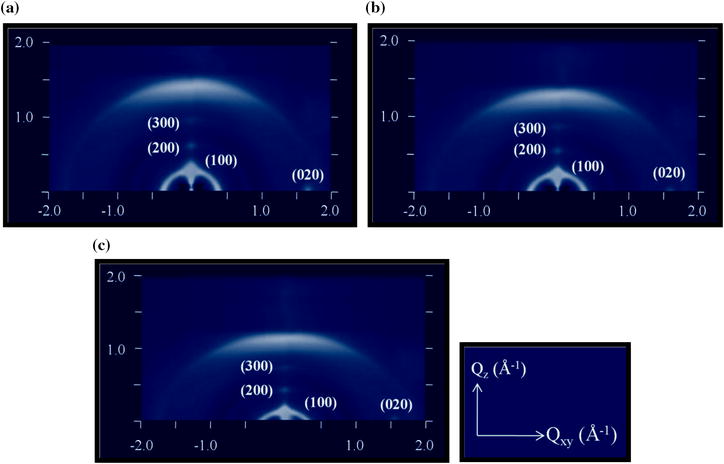
Furthermore, as shown in the two-dimensional grazing incidence wide-angle X-ray scattering (2D-GIWAXS) plots of Fig.
3
a–c, the (100) growth planes (hexyl side chains direction) and (020) growth planes (π–π stacking direction) appeared in out of plane (OOP) and in plane (IP) axes, respectively. In different parts of Fig.
3
, the 2D GIWAXS plots of P3HT
7150
single crystals grown from toluene (
T
s
= 20 °C,
T
c
= 0 °C), xylene (
T
s
= 40 °C,
T
c
= 10 °C), and anisole (
T
s
= 50 °C,
T
c
= 40 °C) are reported.
The 2D GIWAXS plots of P3HT
7150
single crystals grown from toluene at
T
s
= 20 °C,
T
c
= 0 °C (
a
), xylene at
T
s
= 40 °C,
T
c
= 10 °C (
b
), and anisole at
T
s
= 50 °C,
T
c
= 40 °C (
c
)Fig. 3
In the investigated growth systems, the P3HT chains with the lower molecular weights (=7150 and 21,000 g/mol) depicted backbone laminating states; however, those with M n P3HT of 48,800 g/mol were folded in the c -direction. The folding numbers for the homopolymer single crystals were obtained by dividing the P3HT extended length to the width of single crystals (i.e., D (002) dimension detected from width of AFM height profiles). In a similar growth condition, better solvents induced a higher number of foldings. The folding number mostly reduced parallel with T c enhancement. In toluene at T s = 30 °C, by increasing T c from 0 to 10 and 20 °C, the foldings in P3HT 48800 single crystals changed from 3 to 2 and finally 1. The maximum P3HT backbone lamination of 60 occurred in P3HT 7150 single crystals grown from anisole (the weakest utilized solvent in this work) at T s = 60 °C and T c = 20 °C. By increasing M n P3HT from 7150 to 21 000 g/mol, a fewer numbers of the P3HT backbones were laminated. With enhancing the crystallization temperature, the driving force for backbone laminating decreased for homopolymer single crystals. In P3HT 7150 single crystals grown from xylene at T s = 40 °C, through altering T c from 10 to 20 and 30 °C, the backbone laminating numbers were equal to 25, 22, and 18, respectively. The weaker solvents caused P3HT 7150 and P3HT 21000 chains to have more backbone laminations. In P3HT 7150 single crystals grown at T s = 30 °C and T c = 10 °C, the laminating numbers in toluene and xylene were 6 and 25, respectively. By increasing the molecular weight of P3HT block from 7150 to 21,000 g/mol in a similar growth condition, the laminating numbers changed to 3 and 10 in toluene and xylene, respectively.
By increasing the molecular weight of P3HT, the growth of single crystal occurred slowly in the a -direction. In anisole at T s = 50 °C and T c = 20 °C, the thicknesses of P3HT 7150 and P3HT 21000 single crystals were 1180.22 and 1130.14 nm, respectively. The poorer solvent resulted in thicker fibrillar single crystals. The thicknesses of fibrillar P3HT 21000 single crystals in toluene and xylene were 95.00 and 401.00 nm, respectively. The elevated crystallization temperatures due to providing a lower driving force for stacking of the P3HT chains in the hexyl chains direction led to thinner fibrils. In xylene at T s = 30 °C, the thicknesses of P3HT 48800 single crystals grown at the crystallization temperatures of 10 and 20 °C were 404.56 and 365.70 nm, respectively. The stacking also increased in π–π direction using the poorer solvents. At T s = 30 °C and at T c = 10 °C, the lengths of P3HT 48800 single crystals were 34.85 and 54.02 µm in toluene and xylene, respectively. Via M n P3HT increase, π–π stacking was weakened in the fibrillar single crystals. In xylene at T s = 30 °C, the lengths of P3HT 7150 , P3HT 21000 , and P3HT 4880 single crystals were 59.75, 56.79, and 53.89 µm, respectively.
Rectangular/hexagonal dielectric PEG single crystals
The PEG single crystals developed from dilute solution were rectangular; however, the corresponding single crystals grown from melt state were hexagonal. It could be ascribed to the regular prism faces which are (120) and (140) for solution- and melt-grown single crystals, respectively. In general, the growth conditions led to the appearance of growth prisms with different growth rates. By competing with the growth planes having different rates, the prisms with the lower growth rates dictated the ultimate lateral habit or shape of developed single crystals. The seeds acquired from the self-seeding step were also analyzed. In either solution or melt circumstance, the size of seeds ranged 0.5–1 nm. The shape of seeds was similar and cubic-like in solution and melt states. Therefore, this was not the type or shape of added seeds which determined the final lateral habits of grown single crystals. The subsequent growth condition such as the concentration governed the lateral habits and lateral sizes.
AFM height images of homo-PEG
5000
single crystals grown from amyl acetate and molten thin film at
T
c
= 28 °C accompanied by the SAED patterns and AFM height profiles are illustrated in Fig.
4
a, b. The thickness of the hexagonal single crystal grown from melt was 16.3 nm while that of solution-grown one was 11.1 nm. This difference was also detected for the lateral sizes. The lateral size of melt-grown single crystals was 24.5 μm; however, that of solution-grown one was 6.5 μm. Generally, the feasible prism fronts for a PEG single crystal was categorized as (110), (120), (140), (100), (200), (010), (020), and (040) [
42
]. As mentioned above, in lamellar growth of single crystal, different prism fronts are in competition with each other, and those that have lower rate could form the determinant faces because from a kinetic point of view, lower rate is a controlling factor. The dilution of system as well as strength of interactions before being included in the crystalline structure could in turn affect the growth fronts. Furthermore, both solution-grown and melt-grown single crystals demonstrated a flat-on orientation, in which the lamellar PEG backbones were perpendicular to the substrate (Fig.
5
), via two symmetric pairs of (120) and (140) spots in electron diffraction patterns.
TEM image (
top
) and the corresponding ED pattern in the inset as well as AFM height image (
bottom
,
left
) and height profile (bottom, right) of homo-PEG
5000
single crystals at
T
c
= 28 °C;
a
solution state (amyl acetate), thickness: 11.1 nm and lateral size: 6.5 μm;
b
melt state, thickness: 16.3 nm and lateral size: 24.5 μm Schematic representation of lamellar PEG single crystals prepared by homo-PEG and PEG block copolymers attached to a coily amorphous chainFig. 4
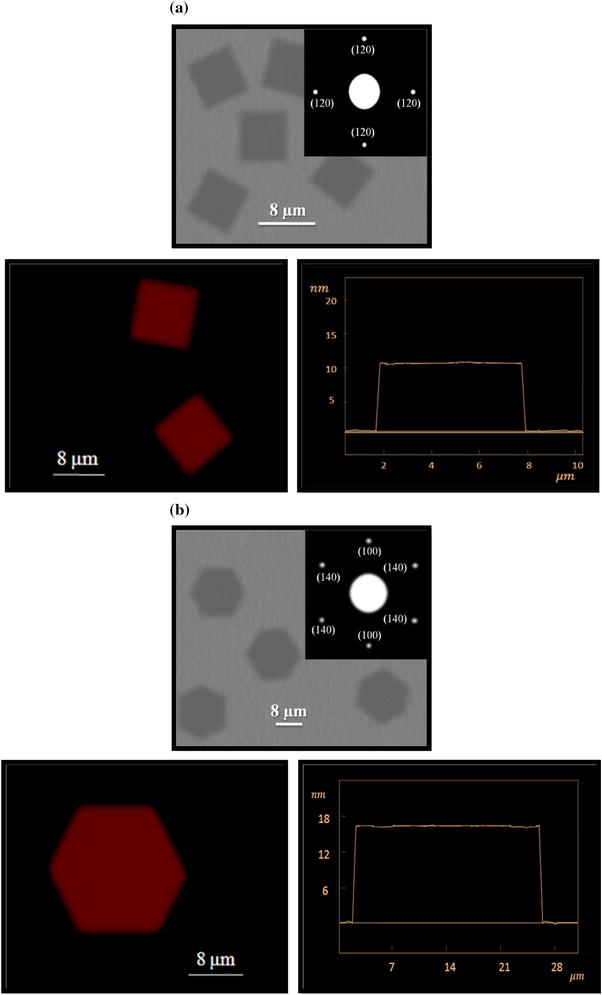
Fig. 5
Totally, as mentioned in the previous section, when the comb-like conjugated backbones such as P3HT assembled to construct an ordered crystalline structure, two different types of stackings comprising π–π stacking in the longitude of fibrillar crystals or in the (020) axis and also the hexyl side chains stacking in the thickness of fibrils or in the (100) axis occurred. Based on dimensions of fibrillar single crystals in the a , b , and c directions, the edge-on and flat-on orientations were detected. On the other side, for the PEG lamellar single crystals usually a flat-on orientation with (120) prisms in dilute solution state and (100) and (140) prisms in the melt state was detected. Moreover, the conjugated P3TH chains because of having a rigid nature had a tendency to possess the backbone foldings in the higher molecular weights, here 48800 g/mol, compared to the coily PEG backbones. Actually, the PEG blocks were capable of folding even in very low molecular weights, i.e., 5000 g/mol. In contrast, the P3HT backbones up to 21,000 g/mol were laminated on each other (Fig. 2 bottom), and chain folding was only detected for P3HTs having the molecular weight of 48,800 g/mol (Fig. 2 top).
The 2D small-angle X-ray scattering (SAXS) pattern in Fig.
6
a is indicative of two sorts of scattering which are dense polymers such as discrete interferences and diffuse central section. The diffuse scattering may originate from the micro-voids of the sample. The micro-voids are unfilled spaces between the lamellae [
52
,
53
]. Discrete interferences are shown on the meridian of the 2D SAXS pattern. A periodic structure could be inferred from orders of the main interference maximum. A typical measurement performed on single crystal mat of homo-PEG
5000
developed at 28 °C is given in Fig.
6
b. In 2D SAXS pattern, the scattering intensity was concentrated in the equatorial plane, and it depicted at least three orders of the interference maximum proving good ordering within the single crystal stacks. In the ideal case of stacked parallel platelets, the pattern should contain only a series of equatorial spots.
2D SAXS pattern (
a
), reduced scattering curve (
b
) and 1D correlation function (
c
)Fig. 6
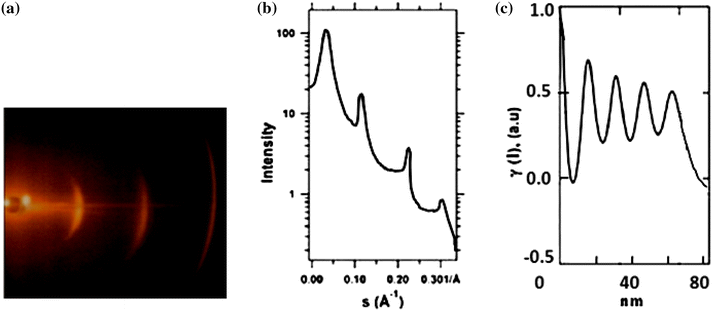
Figure
6
c shows 1D correlation function (γ) presenting the micro-structural parameters. This graph represented that the sample had an ideal lamellar morphology as shown in Fig.
5
. First flat minimum of 1D correlation function was used to determine the value of crystallinity (Φ
L
). The long period or thickness referred as the Bragg peak was predicted from the position of the first maximum of 1D correlation function (γ). The γ
min
was related to the crystallinity, Φ
L
, as followed by Eq. (
1
) [
54
].
Here, γ min = −0.05; so, Φ L = 0.95 or 95%. The AFM height profiles and SAXS 1D correlation functions had a high consistency with each other. For homo-PEG 5000 single crystals grown from melt state, the average thickness from AFM height profile (Fig. 4 b) and SAXS 1D correlation functions (Fig. 6 c) was equal to 16.3 and 16.9 nm, respectively.
In melt growth systems of homopolymers, population of non-ideal structures was high in comparison to solution growth systems from which we observed pyramids with hexagonal bases. As a fact, the overall principle of effect of crystallization temperature on crystal thickness was not satisfied for non-ideal crystals while lateral size of such structures was highly compatible with that of ideal and monolayer single crystals (with error percentage below 5%). Likewise, in dilute solution systems prepared with amyl acetate, the pyramids had square bases. In some cases, the conic crystals were detectable as well. Some off-central pyramids and cones were recognized in which the upper layer had deviation from bottom layers. Figure
7
a depicts TEM images of crystals in question. In our work, the lens-shaped crystals were also observed for the homo-PEG
5000
. The lens-shaped crystals were previously reported for PS-
b
-PEO by Zheng and co-authors [
55
].
TEM images of off-central conic crystal in solution growth system (
up
) and pyramidal crystal in melt growth system (
down
) of homo-PEG
5000
; the
scale bar
is 3 µm for solution state and 11 µm for melt state (
a
); AFM height image of PEG
5000
-
b
-PS
14800
/PEG
5000
random single-co-crystals grown from amyl acetate at
T
c
= 28 °C showing the uniformity in size and shape (
b
); STEM image of PEG
5000
-
b
-PMMA
13100
/PEG
5000
random single-co-crystals grown from amyl acetate at
T
c
= 23 °C (
c
); the weight ratio of PEG
5000
-
b
-PS
14800
/PEG
5000
and PEG
5000
-
b
-PMMA
13100
/PEG
5000
was 50/50 wt/wt for co-crystallizationFig. 7

The effect of self-seeding temperature on single crystal lateral size was investigated in the frame of several experiments. For solution-grown single crystals of homo-PEG 5000 (in amyl acetate) when self-seeding temperature changed from 39 to 41 °C, the lateral size varied from 4.38 to 6.54 μm. This indicated that proportional to elevation of self-seeding temperature, the lateral size of single crystals increased while their population in the system decreased. More details are reported elsewhere [ 56 ]. The crystallization temperature had the same effect on the thickness of developed single crystals.
Homopolymer/block copolymer single-co-crystals
Before focusing on details, we are supposed to bring some notions into consideration. A random single-co-crystal is a uniform crystalline structure having two or more crystallizable constituents. The surface morphology of these single crystals resembled homo-brush single crystals. Figure 7 b, c depicts AFM height images of PEG 5000 - b -PS 14800 /PEG 5000 random single-co-crystals grown at T c = 28 °C and scanning transmission electron microscopy (STEM) image of PEG 5000 - b -PMMA 13100 /PEG 5000 random single-co-crystals grown at T c = 23 °C, respectively.
In the current paper, two approaches were presented for verification of random single-co-crystals. First, the total thickness obtained by AFM was higher for random co-crystals in comparison to corresponding homo-brush single crystals. The height profiles of the three sorts of single crystals crystallized at T c = 23, 28 and 32 °C in dilute solution of amyl acetate including homo-PEG 5000 (10.33, 11.09 and 11.55 nm), random co-crystal of PEG 5000 - b -PS 4600 /PEG 5000 (16.12, 17.19 and 18.61 nm) and homo-brush of PEG 5000 - b -PS 4600 (13.96, 15.12 and 16.65 nm) proved this claim. The mentioned trend was also detected in PMMA-based systems. The total thickness of single crystals grown at the same temperatures for homo-PEG 5000 , random co-crystal of PEG 5000 - b -PMMA 13100 /PEG 5000 , and homo-brush of PEG 5000 - b -PMMA 13100 were 10.33, 15.52 and 11.33 nm, respectively. Irrespective of crystallization temperature and the sort of amorphous block, via increasing the amorphous block molecular weight, due to elevation of demanded coverage surface area and thus a bigger number of folds of the PEG crystal, the osmotic pressure on the substrate increased, thereby its thickness decreased. In addition, for all samples of the same group, through elevating the crystallization temperature, the increase of fold-length was a consequence of the system tendency towards the lowest state of free energy, which belonged to the fully extended chain crystals [ 18 , 54 ].
Second, comparing TEM electron diffraction (ED) patterns of homo-PEG and homo-brush single crystals, one could behold that the central sphere of homo-brush single crystal is smaller than that of homopolymer crystal (Fig.
8
). It could be related to higher order of homopolymer single crystals which did not have any disturbing amorphous brushes in their structure. Similarly, at the same growth condition, the size of central sphere for random co-crystals was greater in comparison with corresponding homo-brush single crystals, whereas it was smaller than that of respective homopolymer single crystals (Fig.
8
). Due to presence of homo-PEG chains which did not have any amorphous blocks in random single-co-crystal structure, its ordering was higher than that of corresponding homo-brush single crystal. Hence, the central spot of random single-co-crystal was bigger than that of homo-brush one. On the other side, the structural order of homopolymer single crystals was in a high level in comparison to the random co-crystals. Therefore, the mentioned spot was smaller for the latter ones. TEM bright images and ED patterns for grown single crystals in amyl acetate at
T
c
= 23 °C consisting homo-PEG
5000
, random single-co-crystal of PEG
5000
-
b
-PS
10000
/PEG
5000
and homo-brush of PEG
5000
-
b
-PS
10000
are depicted in Fig.
8
a–c, respectively. The ED patterns of all samples possessed four spots entitled (120) around the central core, representing four dominant fronts filled with the polymer chains. These major fronts constituted four sides of square of ideal and monolayer single crystal. This was in accordance with those that have been reported for homo-PEG and copolymer single crystals having PEG as crystalline substrate up to now [
19
,
42
,
57
,
58
].
TEM bright images accompanied by the corresponding SAED patterns of the cubic single crystals grown in amyl acetate at
T
c
= 23 °C; homo-PEG
5000
(
a
); random co-crystal of PEG
5000
-
b
-PS
10000
/PEG
5000
(
b
); homo-brush of PEG
5000
-
b
-PS
10000
(
c
)Fig. 8

Generally, the determinative parameters on the thickness of single crystal substrate were the molecular weights of crystalline and amorphous blocks as well as the crystallization temperature. The fourth effective parameter that we introduced was disapproaching of tethered chains on the substrate. In conclusion, the relation applied for the grafting density of conventional homo-brush single crystals (Eq.
2
) [
55
] was not more usable for the random single-co-crystals.
Equation ( 2 ) was only applicable for the systems, in which all contributing chains in the single crystal structure had the amorphous blocks. On the other hand, we did not know the accurate percentage of homo-PEG chains in random structure and the heterogeneity of their presence in different parts of single crystal structure as well. So, we were not capable of introducing a comprehensive relation with more scrutiny for grafting density of random single-co-crystal. The higher percentage of homo-PEG chains were included in the single crystal structure, the higher disapproach of tethered chains occurred on the substrate surface. Moreover, with entering the homo-PEG chains in the random structure, the tethered chains on the substrate disapproached. Therefore, the repulsive interaction between the brushes decreases and, consequently, each chain tended to reach more coily conformation. It in turn may cause the amorphous tethered brushes to decrease their height.
Despite considering explanations presented above, if we use the same equation applied for conventional homo-brush single crystals (Eq.
3
) to determine the thickness of crystalline substrate (
d
CRYST
), and subtract it from total thickness (
d
total
) obtained from AFM to gain the amorphous brushes height (
d
AMORPH
) for random single-co-crystals (in comparison with respective homo-brush ones), we see that
d
AMORPH
has risen instead of decreasing.
For the PEG 5000 - b -PS 4600 /PEG 5000 at T c = 23 °C, by subtracting 2 d AMORPH of PEG 5000 - b -PS 4600 (=2 × 3.63 nm) from d total of PEG 5000 - b -PS 4600 /PEG 5000 (=16.12 nm), we could reach the corresponding thickness of random single-co-crystal (8.86 nm). This value was much closer to the respective thickness of homo-PEG 5000 single crystal (=10.33 nm) in comparison with the substrate thickness of corresponding homo-brush (PEG 5000 - b -PS 4600 ) single crystal (=6.70 nm) at the same condition. Comparing these data may help us conclude that in PEG 5000 - b -PS 4600 , the steric repulsive hindrance has been high enough to hamper the formation of thicker crystal, and the cooperation of homo-PEG chains has been very effective to reduce the osmotic pressure, and cause the substrate to increase its thickness.
For PEG 5000 - b -PMMA 8700 /PEG 5000 at T c = 23 °C, by subtracting 2 d AMORPH of PEG 5000 - b -PMMA 8700 homo-brush single crystal (2 × 3.30 nm) from d total of PEG 5000 - b -PMMA 8700 /PEG 5000 random co-crystal (13.55 nm), we could thus determine the corresponding crystalline substrate thickness for PEG 5000 - b -PMMA 8700 /PEG 5000 (=6.95 nm). This obtained d CRYST was somewhat located in the middle of range of homo-brush single crystal thickness ( d PEG ) and that of PEG 5000 one (3.67–10.33 nm). With increase of the amorphous block molecular weight, the portion of homo-PEG chains in the crystal structure has been raised, and it could be attributed to higher tendency of system to attract the homopolymers to lift the respective osmotic pressure off the substrate surface. Via increasing the amorphous block molecular weight, the critical quorum demanded to lift the osmotic pressure off the substrate, and to heighten its thickness increased. The effective parameters on the hindrance of single crystal systems fell into the solvent quality, the amorphous block molecular weight, and the interaction between the tethered amorphous chains and substrate surface. In these systems, the most dominant parameter on the substrate thickness was interaction between brushes and substrate surface. The mentioned trend was satisfied at elevated crystallization temperature as well.
Comparing the substrate thickness of homo-brush and random single-co-crystal of PEG
5000
-
b
-PS
14800
and PEG
5000
-
b
-PS
14800
/PEG
5000
(4.14 vs. 9.85 nm) (the highest
M
n
PS
) with those of PEG
5000
-
b
-PMMA
8700
and PEG
5000
-
b
-PMMA
8700
/PEG
5000
(3.67 vs. 6.95 nm) (the lowest
M
n
PMMA
) at
T
c
= 23 °C,
d
PEG
in PEG
5000
-
b
-PS
14800
/PEG
5000
was much closer to
d
PEG
in homo-PEG
5000
in comparison to PEG
5000
-
b
-PMMA
8700
/PEG
5000
. Hence, the attractive interaction between PMMA brushes and substrate surface had the most dominant effect on the substrate thickness, and even in the presence of homo-PEG chins it did not allow the substrate to increase its thickness. Different parts of Fig.
9
show the above-mentioned trends.
The trend of d
PEG
variations with
T
c
for PEG
5000
-
b
-PS
14800
/PEG
5000
, PEG
5000
-
b
-PMMA
8700
/PEG
5000
single-co-crystals, PEG
5000
-
b
-PS
14800
, PEG
5000
-
b
-PMMA
8700
and homo-PEG
5000
single crystals (
a
); the trend of thickness variation with
T
c
for PEG
5000
-
b
-PS
4600
/PEG
5000
, PEG
5000
-
b
-PS
4600
and homo-PEG
5000
(
b
), PEG
5000
-
b
-PS
10000
/PEG
5000
, PEG
5000
-
b
-PS
10000
and homo-PEG
5000
(
c
) and PEG
5000
-
b
-PS
14800
/PEG
5000
, PEG
5000
-
b
-PS
14800
and homo-PEG
5000
(
d
) single crystals; the effect of
T
c
(
e
) and
M
n
PMMA
(
f
) on the thickness of PEG
5000
-
b
-PMMA/PEG
5000
single-co-crystals and PEG
5000
-
b
-PMMA single crystalsFig. 9
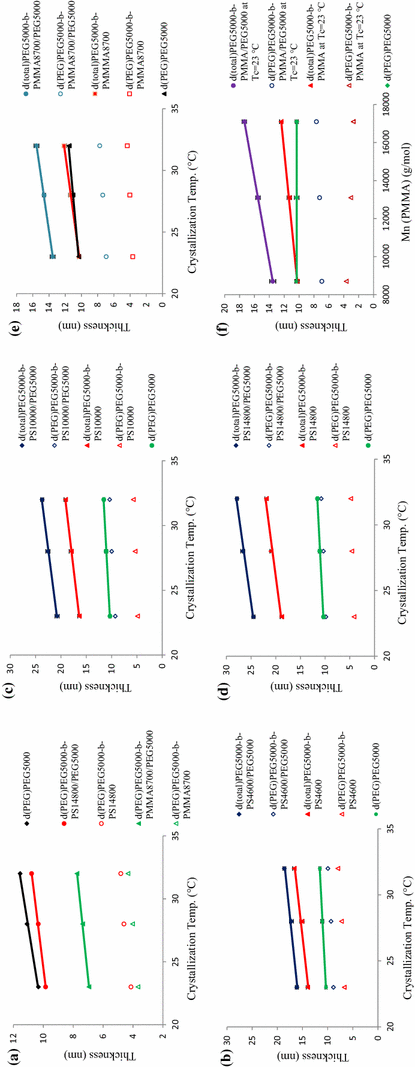
As a fact, via enhancement of amorphous blocks’ molecular weight, especially for the PS, d PEG of random single-co-crystal approached the respective thickness of homo-PEG single crystal grown at the same condition; because by increasing the length of amorphous brushes, the steric repulsion exerted between them increased and, consequently, the presence of homo-PEG chains (after reaching to the critical level) was capable of highly lifting the osmotic pressure off the substrate surface. So, difference between random co-crystal thickness and that of corresponding homo-PEG single crystal diminished. On the other hand, the critical quorum for increase of substrate thickness was higher in samples having longer amorphous chains. That is why to alter d PEG , a larger number of homo-PEG chains should be included into the random single-co-crystal structure. At T c = 23 °C for PEG 5000 - b -PS 4600 /PEG 5000 , PEG 5000 - b -PS 10000 /PEG 5000 and PEG 5000 - b -PS 14800 /PEG 5000 random co-crystals, d PEG ranged from 8.86 to 9.28 nm and, subsequently, to 9.85 nm, respectively. By increasing the M n PS from 4600 to 14800 g/mol, d PEG of random single-co-crystals reached d PEG of homo-PEG 5000 . The d PEG s of homo-brush single crystals decreased and disapproached from d PEG of homo-PEG 5000 . Different parts of Fig. 9 show the above-mentioned trends.