Graphical Abstract

First-principle calculations within density functional theory were performed to investigate the interactions of NO and NO 2 molecules with TiO 2 /MoS 2 nanocomposites. Given the need to further comprehend the behavior of the NO x molecules positioned between the TiO 2 nanoparticle and MoS 2 monolayer, we have geometrically optimized the complex systems consisting of the NO x molecule oriented at appropriate positions between the nanoparticle and MoS 2 monolayer. The structural properties, such as bond lengths, bond angles, adsorption energies and Mulliken population analysis, and the electronic properties, including the density of states and molecular orbitals, were also analyzed in detail. The results indicate that the interactions between NO x molecules and N-doped TiO 2 in TiO 2 -N/MoS 2 nanocomposites are stronger than those between gas molecules and undoped TiO 2 in TiO 2 /MoS 2 nanocomposites, which reveal that the N-doping helps to strengthen the interaction of toxic gas molecules with hybrid TiO 2 /MoS 2 nanocomposites. The N-doped TiO 2 /MoS 2 nanocomposites have higher sensing capabilities than the undoped ones, and the interaction of NO x molecules with N-doped nanocomposites is more favorable in energy than the interaction with undoped nanocomposites. Therefore, the obtained results also present a theoretical basis for the potential application of TiO 2 /MoS 2 nanocomposite as an extremely sensitive gas sensor for NO and NO 2 molecules.
Titanium dioxide (TiO 2 , Titania) has aroused great attentions as an important semiconductor material due to its effectiveness and outstanding properties, such as non-toxicity, low cost, high catalytic efficiency, photoactivity [ 1 ], and stability. TiO 2 has been widely utilized in many fields, such as photo-catalysis, gas sensing, organic dye-sensitized solar cells, water splitting, and pollutant degradation [ 2 – 5 ]. Three important polymorphs were found for TiO 2, namely, anatase, rutile, and brookite [ 6 ], in which anatase and rutile forms are the most widely studied ones in different fields of science and technology. The photocatalytic applications of TiO 2 were restricted due to its wide bandgap (3.2 eV), which allows the absorption of the solar spectrum at the ultraviolet region by a lower percentage (3–5 % of the incoming solar light). The doping of TiO 2 anatase with some nonmetal elements, such as nitrogen, is a convenient solution, which would enhance the photo-efficiency of TiO 2 to the visible region and improve its photocatalytic activity [ 7 , 8 ]. Two-dimensional (2D) semiconductor materials, such as MoS 2 [ 9 ], and other dichalcogenides consisting of transition metals, such as MoSe 2 , WS 2, and so on, indicate the final scale for chalcogenide dimension around the vertical direction. MoS 2 , a layered structure consisting of Mo and S atoms arranged in hexagonal structure of atomic sheets of molybdenum and sulphur atoms, attracts numerous attentions due to the excellent electrical, mechanical, and optical properties, such as satisfied bandgap, thermal stability, carrier mobility, and so on [ 10 – 12 ]. MoS 2 has been broadly used in abundant applications, such as photocatalysts, nanotribology, lithium battery, dry lubrication, hydrodesulfurization catalyst, and photovoltaic cell because of its unique electronic, photosensitive, and catalytic properties [ 13 – 16 ]. Nanoelectronic devices fabricated on 2D materials, such as MoS 2, suggest also many efficiencies for these layered materials, which cause the further miniaturization of the integrated circuits beyond Moore’s law. Recently, numerous electronic devices were made using the few-layer MoS 2 as an important component, such as field-effect transistors [ 17 ], sensors [ 18 ], etc. However, several computational studies of N-doped TiO 2 anatase nanoparticles and few-layer MoS 2 structures have been separately published, describing some of the main electronic and physical properties of these materials. Particularly, the gas-sensing capabilities of MoS 2 -based field-effect transistors and sensing films for NO and NH 3 were experimentally revealed with an enhanced sensitivity in some other works [ 19 , 20 ]. TiO 2 /MoS 2 nanocomposites have been successfully synthesized for different purposes by some experimental methods [ 21 – 23 ]. There are no explanative computational studies on the adsorption behaviors of TiO 2 /MoS 2 nanocomposites. NO x molecules have been characterized as toxic gases which are mainly emitted from power plants and vehicle engines. For the general public, the most outstanding provenance of NO 2 is internal combustion engines that burn fossil fuels to work properly. In indoor places, NO 2 emission mainly stems from cigarette smoke, kerosene heaters, and stoves. Therefore, optimal removal of these harmful molecules is an important subject to human health and environmental protection [ 24 ]. In this study, the interaction of NO x molecules with TiO 2 /MoS 2 nanocomposites has been investigated by density functional theory (DFT) computations. We present here the results of calculations of complex systems consisting of NO x molecule positioned between the TiO 2 anatase nanoparticle and MoS 2 monolayer. The electronic structure of the adsorption systems has also been analyzed, including the projected density of states (PDOSs) and molecular orbitals (MOs). The main aim of this study is to supply an overall understanding on the adsorption behaviors of nano-TiO 2 /MoS 2 composites as highly sensitive NO x sensors.
DFT calculations [
25
,
26
] were performed as implemented in the Open-Source Package for Material eXplorer (OPENMX3.8) [
27
], being a well-organized software package for nano-scale materials simulations based on DFT, PAO basis functions, and VPS pseudopotentials [
28
,
29
]. Pseudo-atomic orbitals were utilized as basis sets in the geometry optimizations. The considered cut-off energy is set to the value of 150 Rydberg in our calculations [
29
], The PAOs are generated via the basis sets (3-s, 3-p, and 1-d) for Ti atom, (3-s, 3-p, and 2-d) for Mo atom, (2-s and 2-p) for O and N atoms, (3-s and 3-p) for S atom with the chosen cut-off radii of 7 for Ti, 9 for Mo, 5 for O and N, and 8 for S (all in Bohrs). The generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) was used to describe the exchange–correlation energy functional [
30
]. The convergence criterion of self-consistent field calculations was set at 1.0 × 10
−6
Hartree, whereas that of energy calculation was chosen to be 1.0 × 10
−4
Hartree/bohr. For the geometry optimization, ‘Opt’ is used as the geometry optimizer, which is a robust and efficient scheme. The crystalline and molecular structure visualization program, XCrysDen [
31
], was employed for displaying molecular orbital isosurfaces. The box considered in these computations contains 96 atoms (24 Ti, 48 O, 8 Mo, and 16 S atoms) of undoped or N-doped TiO
2
nanoparticle with MoS
2
monolayer. The Gaussian broadening method for evaluating electronic DOS is used. For NO
x
adsorption on the TiO
2
/MoS
2
nanocomposite, the adsorption energy is computed via the following formula:
The chemical formulae of nitric oxide and nitrogen dioxide molecules are NO and NO
2
. NO has linear structure, while NO
2
represents a bent geometrical structure. The structures of NO and NO
2
molecules were represented in Fig.
1
. Distances and angles of the considered molecules were computed in a large cubic supercell. The calculated N–O bond length of free NO molecule is 1.16 Å, while for the bent structure of NO
2
molecule, the bond length and bond angles were calculated to be 1.20 Å and 134°, respectively. All these computed values are in comprehensive agreement with the computational results and the experimentally reported data [
32
].
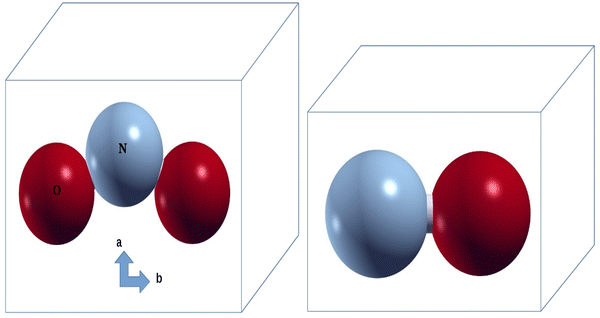
Representation of NO and NO
2
molecules in a large cubic supercellFig. 1
Molybdenum disulfide (MoS
2
) is a layered structure containing molybdenum transition metal, which belongs to the family of two-dimensional dichalcogenides. A hexagonally arrangement of atomic sheets of MoS
2
containing Mo and S atoms set as an S–Mo–S sandwich forms MoS
2
monolayer. The monolayer of MoS
2
model studied here contains 24 atoms in total (8 Mo and 16 S atoms). MoS
2
structure is relaxed for calculating the optimized structural parameters. The calculated S–Mo bond length, Mo–Mo distance, and S–S distance in monolayer are 2.43, 3.20, and 3.15 Å, respectively. These computed bond lengths are somewhat consistent with the values of bulk material [
33
], in reasonable agreement with the reported data [
34
,
35
]. However, there are negligible discrepancies between the results of MoS
2
and its bulk material, which can be ignored. The area of the MoS
2
slab is 9.25 Å × 6.33 Å. The optimized structure of MoS
2
model was displayed in Fig.
2
.
Optimized structure of the chosen MoS
2
monolayer with area values,
a
front view and
b
lateral view. Mo atoms are sketched by
gray balls
and S atoms by
yellow ballsFig. 2

A 3 × 2 × 1 supercell of TiO
2
anatase along x, y, and z directions was utilized for constructing the considered TiO
2
anatase nanoparticles containing 72 atoms. The unit cell is available at “American Mineralogists Database” webpage [
36
] and was reported by Wyckoff [
37
]. Two appropriate oxygen atoms of TiO
2
nanoparticle were replaced by nitrogen atoms to model the N-doped particles. Doping of TiO
2
nanoparticle with nitrogen atom is done according to two doping positions. These two doping positions refer to the middle oxygen and twofold coordinated oxygen atoms substitutions illustrated by O
C
and O
T
in Fig.
3
a, respectively. The area of the anatase nanoparticle is 13.03 Å × 8.24 Å. The crystalline structure of TiO
2
contains two kinds of titanium atoms, namely, fivefold coordinated titanium (5f-Ti) and sixfold coordinated one (6f-Ti), and two kinds of oxygen atoms, namely, threefold coordinated oxygen (3f-O) and twofold coordinated one (2f-O) atoms (see Fig.
3
a). [
38
] It was found that the twofold coordinated oxygen and fivefold coordinated titanium atoms are more reactive than the threefold coordinated oxygen and sixfold coordinated titanium atoms due to the undercoordination in twofold coordinated oxygen and fivefold coordinated titanium atoms. The thickness of the vacuum spacing is 11.5 Å, which is helpful to avoid the additional interactions between the neighbor particles. The optimized structure of TiO
2
/MoS
2
nanocomposite was displayed in Fig.
3
b. Figure
4
also displays the optimized geometries of the N-doped TiO
2
/MoS
2
nanocomposites. The results of geometrical optimizations represent that the O
T
-substituted TiO
2
/MoS
2
nanocomposite is more favorable in energy than the O
C
-substituted one.
Optimized structures of
a
undoped 72 atom TiO
2
anatase nanoparticle constructed using the 3 × 2 × 1 unit cells (O
C
: central oxygen; O
T
: twofold coordinated oxygen; O
D
: dangling oxygen) and
b
TiO
2
/MoS
2
nanocomposite constructed from TiO
2
nanoparticle and MoS
2
monolayer. Ti atoms are sketched by
dark gray balls
, O atoms by
red balls
and N atoms by
blue balls Optimized geometries of two types of N-doped TiO
2
/MoS
2
nanocomposites.
a
O
C
-substituted nanocomposite, which refers to substitution of a threefold coordinated oxygen atom and
b
O
T
-substituted nanocomposite, representing the substitution of a twofold coordinated oxygen atomFig. 3

Fig. 4

For NO molecule, three adsorption configurations are studied here, including the adsorption configurations of types A, B, and C, as shown in Fig.
5
. The calculated adsorption energy values for NO
x
molecule adsorbed on the considered nanocomposites have been listed in Table
1
. This figure presents the orientations of NO molecule towards the N-doped TiO
2
/MoS
2
nanocomposites. For instance, complex A was made from the TiO
2
/MoS
2
nanocomposite with O
C
-substituted TiO
2
nanoparticle and NO molecule with nearly downward oxygen. NO can stably adsorb on N-doped TiO
2
/MoS
2
nanocomposite, compared to the adsorption on the undoped one. The nitrogen atom of NO molecule is preferentially attracted to the doped nitrogen atom of nanocomposite, resulting in the formation of chemical bond between these two nitrogen atoms. The adsorption energy of the NO molecule on the N-doped TiO
2
/MoS
2
nanocomposite (composite A or B) is much higher than that of undoped TiO
2
/MoS
2
nanocomposite, which reveals that NO molecule has a stronger interaction with N-doped nanocomposite than with undoped one. These results also indicate that the interaction of NO molecules with N-doped TiO
2
/MoS
2
nanocomposites is more energetically favorable than the interaction of NO with undoped nanocomposites. This implies that the N-doped nanocomposite adsorbs NO molecule more effectively compared to the undoped one. Besides this, the configuration B is the most stable configuration compared to the configuration A due to its more negative adsorption energy. Configuration B contains O
T
-substituted nanocomposite with adsorbed NO molecule. The adsorption energy of this configuration is more negative than that of other configurations, which suggests that the adsorption of NO molecule on the O
T
-substituted nanocomposite is more energy favorable than the adsorption on the O
C
-substituted one (see Table
1
). Since a greater value of adsorption energy gives rise to a strong interaction between adsorbate and the adsorbent, it can be seen that there is a stronger interaction between NO and N-doped nanocomposite compared to NO and undoped nanocomposite, implying the dominant effect of N-doping. It means that the nitrogen doping strengthens the interaction of NO with TiO
2
/MoS
2
nanocomposites. The greater the adsorption energy, the higher tendency for adsorption, and, therefore, more efficient adsorption. Table
1
summarizes the bond length values before and after the adsorption of NO molecule on the nanocomposites. The bond lengths given in this table are included N–O bonds of NO
x
molecule, average Ti–N distance, and new N–N and N–O distances between the nanocomposite and adsorbed NOx molecule. The values reported in this table show that the Ti–N bonds and N–O bond of the adsorbed NO molecule are elongated, because the electronic density transfers from the Ti–N bonds of N-doped TiO
2
and N–O bond of the adsorbed NO molecule to the newly formed N–N and N–O distances between the nanocomposite and molecule. This transfer of electronic density indicates that the N–O bond of NO molecule is weakened after the adsorption.
Optimized geometry configurations of the interaction of NO and NO
2
molecules with TiO
2
/MoS
2
nanocomposites. The NO molecule is preferentially adsorbed on the doped nitrogen site of TiO
2
nanoparticle, whereas NO
2
is adsorbed on both the doped nitrogen atom and fivefold coordinated titanium atoms. Configurations
A
–
C
represent the interaction of nanocomposites with NO molecules and the configurations
D
–
I
show the interaction between nanocomposites and NO
2
molecules Bond lengths (in Å), Mulliken charges, and adsorption energies (in eV) for NO
x
molecule adsorbed on TiO
2
/MoS
2
nanocomposites Bond N–O1 bond length N–O2 bond length New N–N bond length Average Ti–N length New N–O bond length Mulliken charge Adsorption energy Composite NO adsorption Non-adsorbed 1.16 – – 1.84 1.94 – −0.122 −2.18 a 1.23 1.63 2.12 – b 1.30 – 1.43 1.99 – −0.066 −3.01 c 1.19 – – – 3.16 −0.269 −0.50 NO2 adsorption Non-adsorbed 1.20 1.20 – 1.84 1.94 d 1.31 1.30 1.45 2.18 −0.064 −1.43 e 1.30 1.30 1.48 2.01 −0.107 −2.12 f 1.28 1.28 – – 2.50 −0.014 −0.22Fig. 5

Table 1
The interaction of NO
2
molecule with the substituted nitrogen atom of TiO
2
/MoS
2
nanocomposites has also been displayed in Fig.
5
as specified by types D-F adsorption geometries. For the doped nitrogen site, the adsorption process is expected to be more energy favorable than that on the dangling oxygen atom site. The reason can be simply sought using the data collected in Table
1
. Similarly, the NO
2
molecule preferentially interacts with the doped nitrogen site on the nanoparticle surface, in comparison with the other surface oxygen atoms. Table
1
also lists the lengths for Ti–N bonds and N–O bonds of the adsorbed NO
2
molecule and the newly formed N–N and N–O distances. The results of this table indicate that the Ti–N bonds of the nanocomposite and the N–O bonds of the NO
2
molecule are stretched after NO
2
adsorption. Because the electronic density transfers from the Ti–N bonds and N–O bonds to the newly formed distances between the nanocomposite and adsorbed NO
2
molecule. In configuration D, the nitrogen atom in the NO
2
molecule interacts with the doped nitrogen site on the TiO
2
nanoparticle to form a strong chemical bond and, therefore, strong interaction (1.45 Å N–N bond length). Among three models for NO adsorption, the adsorption configuration in which the nitrogen atom of NO interacts with the doped nitrogen site of TiO
2
at O
T
position is the most energy favorable one. In the case of NO
2
adsorption, the adsorption energy of configuration E is much higher (more negative) than that of configuration D and undoped system adsorption (configuration F). It should be noted that the adsorption on the O
T
-substituted nanocomposite leads to the stable configurations (stronger interactions), compared to the adsorptions on the O
C
-substituted one. As can be seen from Tables
1
and
2
, the adsorption energies on the O
T
-substituted nanocomposites (complexes E and H) are more negative than the adsorption energies on the O
C
-substituted ones, implying that NO
2
adsorption on the O
T
-substituted nanocomposites is energetically more favorable than the adsorption on the O
C
-substituted ones. As a result, the adsorption of NO
2
molecule on the N-doped TiO
2
/MoS
2
nanocomposite is more energy favorable than the adsorption of NO
2
on the undoped nanocomposite, indicating that the N-doped nanocomposite can adsorb NO
2
molecule more efficiently. Thus, the N-doping strengthens the interaction of NO
2
molecule with TiO
2
/MoS
2
nanocomposites. The O–N–O bond angles of the NO
2
molecule have been decreased after the adsorption process because of the formation of new chemical bond between nitrogen atom of NO
2
with nitrogen atom of TiO
2
nanoparticle. This chemical bond formation leads to an increase in the p characteristics of bonding molecular orbitals. Thus, the
sp
hybridization of nitrogen in the NO
2
molecule converts to near-
sp
3
hybridization. For the case of TiO
2
adsorption on the fivefold coordinated titanium site presenting two contacting point between the nanoparticle and NO
2
molecule, the bond length and bond angle values have been reported in Table
2
. We can see two newly formed Ti–O bonds between the titanium atoms of the nanoparticle with oxygen atoms of NO
2
molecule. This adsorption configuration is referred to as bridge configuration, and their complexes were illustrated in Fig.
5
(G–I complexes). The N–O bonds of NO
2
molecule have been lengthened after the adsorption process, suggesting the weakening of the N–O bonds. In addition, the adsorption process results in a decrease in the O–N–O bond angle values of NO
2
. The adsorption energy analysis reveals that the complex H is the most energy favorable complex in comparison with complex G and both are more stable than the pristine system adsorption. These results show that the NO
x
adsorption on the N-doped nanocomposite is more favorable in energy than the NO
x
adsorption on the pristine nanocomposite. By considering this, we found that the nitrogen doping strengthens the interaction of NO
x
molecule with TiO
2
/MoS
2
nanocomposites. The obtained improvements on the structural and electronic properties of TiO
2
/MoS
2
nanocomposites here represent that the N-doped TiO
2
-based nanocomposite can be efficiently utilized in the removal and sensing of toxic NO
x
molecule.
Bond lengths (in Å), angles (in degrees), Mulliken charges, and adsorption energies (in eV) for complexes providing two contacting point between NO
2
molecule TiO
2
/MoS
2
nanocomposites Composite New Ti–O1 bond length New Ti–O2 bond length N–O1 bond length N–O2 bond length O–N–O bond angle Mulliken charge Adsorption energy g 2.41 2.53 1.28 1.29 122.3 −0.101 −1.42 h 2.03 2.28 1.30 1.36 117.4 0.180 −2.22 i 2.14 2.36 1.30 1.34 120.6 0.145 −0.92Table 2
Figure
6
presents the total density of states (TDOS) for N-doped TiO
2
anatase nanoparticles and corresponding TiO
2
/MoS
2
nanocomposites. This figure reveals a creation of small peak in the density of states (DOSs) of N-doped nanocomposite at the energy ranges near to -12 eV. TDOSs of adsorption configurations were also displayed in Fig.
7
. A closer inspection of these figures indicates the increase of the discrepancies between DOS of N-doped TiO
2
and nanocomposite by adding the MoS
2
monolayer and adsorption of NO
x
. These differences included considerable shifts in the energies of the peaks and appearance of some peaks in the DOS of the studied systems. As distinct from these figures, the DOSs of the considered nanocomposites were mainly shifted to the lower energy values after the adsorption process. Therefore, the resultant variations in the energy of the states can have positive effects on the electronic transport properties of the nanocomposites and in turn can provide a helpful procedure for designing and engineering NO
x
sensors based on N-doped TiO
2
and two-dimensional transition metal dichalcogenides (i.e., MoS
2
monolayer). The projected partial density of states (PDOSs) for the interaction of NO
x
molecule with TiO
2
/MoS
2
nanocomposites have been displayed in Fig.
8
a–d. Panels (a, b) present the PDOS of the nitrogen atom of NO molecule and the doped nitrogen atom of N-doped nanocomposite. The large overlap between the PDOS of the mentioned atoms exhibits that the nitrogen atom of NO molecule interacts with the doped nitrogen atom of nanocomposite, suggesting the formation of new N–N bond. The PDOSs for NO
2
adsorption on the doped nitrogen site have also been shown as panels (c, d), which indicate a high overlap between the PDOS of nitrogen atom of NO
2
molecule and the nitrogen atom of nanocomposite and consequently forming a chemical bond. For NO and NO
2
adsorption on the middle oxygen (O
C
site), the calculated PDOSs have been displayed in Fig.
9
(panels a, b), representing a low PDOS overlap between the nitrogen atom of NO and NO
2
molecules and the O
C
atom of nanocomposite. This means a weak interaction between NO
x
and nanocomposite. The other panels of Fig.
9
represent the PDOS of oxygen atom of nanocomposite before and after the adsorption on the undoped nanocomposite, as well as the PDOS of nitrogen atoms of NO and NO
2
molecules. As can be seen, the main difference is the creation a small peak in the PDOS curves and also shifting the position of the peaks to the lower lying energies. Figure
10
a–d shows the PDOS of the nitrogen atom of nanocomposite before and after the adsorption on the N-doped nanocomposite, which also suggests a shifting of the PDOS of nitrogen atom to the lower energy values. To further discover the electronic variations at the adsorption site, the PDOSs of nitrogen atom of NO and NO
2
molecule before and after the adsorption were also presented in Fig.
11
a–d. Similarly, it can be seen from these PDOS plots that the biggest difference is the creation of some small peaks and also the state changing to the smaller energy values. The PDOSs of the complexes providing two contacting point (G, H and I) have also been displayed in Fig.
12
, which indicate a higher overlap between the PDOS of Titanium atoms with two left and right oxygen atoms in all six panels. This means a formation of two chemical bonds between the titanium and oxygen atoms. Figures
13
and
14
represent the PDOSs of nitrogen atoms and their related p orbitals for complex D. As can be seen from these figures, p
3
orbital of the nitrogen atom of nanocomposite and NO
2
represents a considerable overlap with the other atom participating in chemical bond formation. This is an indication of the higher contribution of p
3
orbital in chemical bond in comparison with the other orbitals. The PDOSs of nitrogen atoms and their p orbitals for complex E have also been displayed in Fig.
15
, indicating a high overlap between the PDOS of nitrogen atom with p
1
atomic orbital compared to the other orbitals. The HOMO and LUMO molecular orbitals were also displayed in Figs.
16
and
17
, respectively. A closer inspection reveals that the HOMOs are strongly located on the nanoparticle, whereas the electronic densities in the LUMOs are mainly dominant on the NO
x
molecules. As can be seen from Fig.
17
, the electronic density in the LUMOs seems to be distributed over the NO
x
molecules and on the middle of newly formed bonds. The accumulation of the electronic density at the middle of the newly formed bonds confirms the formation of new bonds and consequently the transfer of electronic density from the Ti–N bonds and N–O bonds to the newly formed bonds. However, the resultant improvements on the electronic properties of TiO
2
/MoS
2
nanocomposites obtained by N-doping here demonstrate that the N-doped TiO
2
/MoS
2
nanocomposites have stronger sensing capabilities than the pristine ones. To fully analyze the NO
x
adsorption on the considered TiO
2
/MoS
2
nanocomposites, the Mulliken population analysis has been conducted to analyze the charge distribution of the atoms and bonds in a complex system. The calculated Mulliken charge values for studied complexes were collected in Tables
1
and
2
. For complex A, NO
x
adsorption induces a considerable charge transfer of about −0.122 e from NO
x
molecule to the nanoparticle, suggesting that NO
x
acts as an acceptor. In other words, the NO
x
molecule receives electros from nanocomposite. This leads to the changes in the conductivity of the system, which would be an efficient property to aid in the design and fabrication of novel sensor devices for nitrogen oxides recognition.
Total density of states for N-doped TiO
2
and two types of N-doped TiO
2
/MoS
2
nanocomposites.
a
O
C
-substituted nanocomposite and
b
O
T
-substituted one DOS for the different adsorption configurations of the NO
x
molecule on the considered TiO
2
/MoS
2
nanocomposites,
a
A complex (NO molecule adsorbed on the O
C
-substituted nanocomposite);
b
D complex (NO
2
molecule adsorbed on the O
C
-substituted nanocomposite);
c
B complex (NO molecule adsorbed on the O
T
-substituted nanocomposite);
d
E complex (NO
2
molecule adsorbed on the O
T
-substituted nanocomposite) PDOS for the interaction of NO
x
molecule with TiO
2
/MoS
2
nanocomposites,
a
A complex;
b
B complex;
c
D complex;
d
E complex PDOS for the interaction of NO
x
molecule with TiO
2
/MoS
2
nanocomposites,
a
C complex (NO molecule adsorbed on the pristine nanocomposite);
b
F complex (NO
2
molecule adsorbed on the pristine nanocomposite);
c
C complex;
d
F complex;
e
C complex;
f
F complex PDOS of the nitrogen atom of nanocomposite before and after the adsorption process,
a
A complex;
b
B complex;
c
D complex;
d
E complex PDOS of the nitrogen atom of NOx molecule before and after the adsorption process,
a
A complex;
b
B complex;
c
D complex;
d
E complex PDOS of the titanium and oxygen atoms in complexes providing two contacting point between the nanocomposite and adsorbed molecule,
a
G complex (NO
2
adsorbed on the O
C
-substituted nanocomposite in a bridge geometry);
b
G complex;
c
H complex (NO
2
adsorbed on the O
T
-substituted nanocomposite in a bridge geometry);
d
H complex;
e
I complex (NO
2
adsorbed on the pristine nanocomposite in a bridge geometry);
f
I complex PDOS of the nitrogen atoms and their related p orbitals after the adsorption process for D complex PDOS of the nitrogen atoms and their related p orbitals after the adsorption process for D complex PDOS of the nitrogen atoms and their related p orbitals after the adsorption process for E complex Isosurfaces of HOMO molecular orbitals for the adsorption of NO and NO
2
molecules on the TiO
2
/MoS
2
nanocomposites, where |0.05| was used as an isovalue of the molecular orbital Isosurfaces of LUMO molecular orbitals for the adsorption of NO and NO
2
molecules on the TiO
2
/MoS
2
nanocomposites, where |0.05| was used as an isovalue of the molecular orbitalFig. 6

Fig. 7

Fig. 8

Fig. 9

Fig. 10

Fig. 11

Fig. 12

Fig. 13

Fig. 14

Fig. 15

Fig. 16
Fig. 17

DFT calculations were conducted to investigate the interaction of NO x molecules with undoped and N-doped TiO 2 /MoS 2 nanocomposites to effectively understand the sensing properties of these nanocomposites in adsorption processes. The bond angles of the adsorbed NO 2 molecule are decreased compared to those in the isolated gas phase NO 2 , which lead to an increase in the p characteristics of bonding molecular orbitals of nitrogen in the NO 2 molecule. The results also suggest that the N-doped nanocomposites have a higher efficiency to interact with harmful NO x molecules in the environment. In other words, the doping of nitrogen atom provides an increased affinity for the TiO 2 /MoS 2 nanocomposites to interact with NO x molecules. The analysis of adsorption energies reveals that the adsorption of NO x molecules on the N-doped TiO 2 /MoS 2 nanocomposites is more favorable in energy than the adsorption of NO x on the undoped ones. The variation in the electronic structure and molecular orbitals induced by N-doping is found to be responsible for the conductivity of the nanocomposite system. Our calculated results, therefore, suggest a theoretical basis for the prospective application of TiO 2 /MoS 2 hybrid nanostructures as gas sensors for important air pollutants, such as NO and NO 2, in the environment.
This work was supported by Azarbaijan Shahid Madani University [217/D/14271].