Results and discussion
Structure and energetics
The optimized structure of B
3
N
3
C
54
along with C
60
is displayed in Fig.
1
. One can note that the structure of B
3
N
3
C
54
closely resembles that of C
60
in which one of C
6
hexagons is replaced by B
3
N
3
, but its symmetry is reduced to
C
3
point group. To get further insights, we have separately optimized C
6
and B
3
N
3
. After optimization, we have found that they are almost similar in structure, also shown in Fig.
1
. Both possess
D
3h
symmetry and equalized bond lengths, 1.358 Å for B–N and 1.327 Å for C–C. The NPA charges on B and N atoms in B
3
N
3
are +1.04
e
and −1.04
e
, respectively, which suggest polarity of BN bonds. In Table
1
, we have collected the bond lengths of hexagon–hexagon and hexagon–pentagon in B
3
N
3
C
54
and C
60
. Evidently, the B–N bond lengths increase to 1.437 Å and 1.478 Å in hexagon–hexagon and hexagon–pentagon edges which indicate that B–N bonds of the substituted fullerene are weaker than those of free B
3
N
3
. Furthermore, the difference of B–N and C–C bond lengths is significantly increased. This may be expected due to redistribution of charges on B and N atoms in B
3
N
3
C
54
. For instance, NPA charges on B and N atoms in B
3
N
3
C
54
are +1.02
e
and −0.88
e
, respectively. One can note that the polarity of BN bonds reduces in B
3
N
3
C
54
which weakens the bond increasing its bond length.
Equilibrium structures of C
60
(
left
) and B
3
N
3
C
54
(
right
) calculated at B3LYP level Main bond lengths (in Å) of B
3
N
3
C
54
and C
60
at B3LYP/6-31G(d) level System (bond length) Hexagon–hexagon Hexagon–pentagon C60 (C–C) 1.395 1.453 B3N3C54 (B–N) 1.437 1.478 (B–C) 1.526 1.526 (N–C) 1.434 1.434Fig. 1
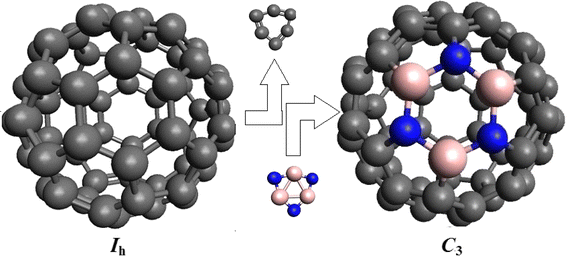
Table 1
The formation of B
3
N
3
C
54
involves two processes: the removal of C
6
hexagon from C
60
and then, inclusion of B
3
N
3
hexagon at the same place. The corresponding stabilization energy can be calculated by,
Spectral properties
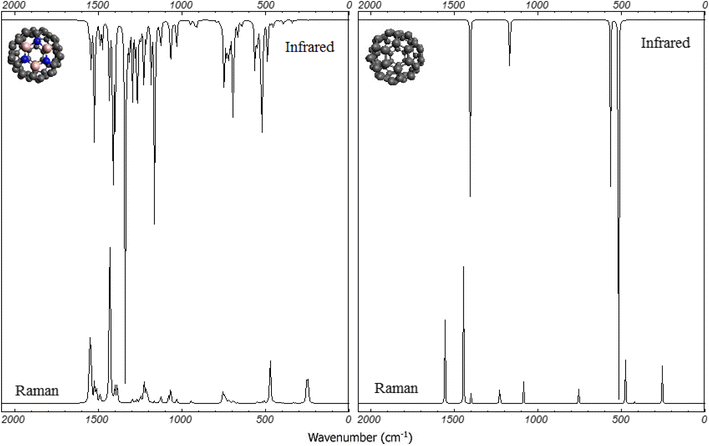
We have calculated vibrational infrared, Raman and NMR spectra of B
3
N
3
C
54
and compared with those of C
60
. The vibrational frequency calculations performed on B
3
N
3
C
54
heterofullerene reveal all real and positive values. This suggests that B
3
N
3
C
54
belongs to at least some local minimum in the potential energy surface. The calculated vibrational infrared and Raman spectra of B
3
N
3
C
54
and C
60
are depicted in Fig.
2
. For
N
-polyatomic systems, there are 3
N
-6 normal modes of vibration. Due to extremely high symmetry of C
60
(
I
h
point group), however, there are only four characteristic modes possessing appreciable intensity. The mode with the highest intensity is assigned at the lowest wavenumber, 537 cm
−1
. The CC stretching modes of C
60
are assigned in the higher wavenumber region, 1460 and 1214 cm
−1
. The calculated wavenumbers were scaled by a factor of 0.9613 as suggested by Merrick et al. [
25
]. The scale wavenumbers of C
60
516, 565, 1167 and 1404 cm
−1
are in accordance with the FT-IR frequencies 526, 576, 1183 and 1429 cm
−1
, respectively, as reported by Chase et al. [
26
]. The substitution of B
3
N
3
into C
60
leads to destruction of the sharpness of peaks in the IR spectrum due to
C
3
point group. As seen in Fig.
2
, a number of additional modes and mixing of various modes appear due to BN, BC and NC stretchings. For instance, the BN stretching modes appear at 1394 cm
−1
. This mode possesses the strongest intensity due to charge transfer from B to N atoms. Furthermore, CC stretching modes of B
3
N
3
C
54
are shifted toward higher wavenumber region as compared to C
60
. For example, the modes calculated at 1606 and 1586 cm
−1
are assigned to CC stretchings with weak intensities. However, CC stretching and lower wavenumber mode of B
3
N
3
C
54
corresponding to C
60
are obtained at 1468 cm
−1
(medium) and 539 cm
−1
(weak). On the contrary, Raman spectrum of B
3
N
3
C
54
closely resembles that of C
60
. There are eight distinct modes of vibrations in the Raman spectra which are in accordance with the experimental reports of Bethune et al. [
27
,
28
]. For instance, the most intense Raman peak calculated at 1465 cm
−1
is observed at 1469 cm
−1
in C
60
solid film [
27
] and 1470 cm
−1
in C
60
thin film [
28
].
Vibrational infrared and Raman spectra of B
3
N
3
C
54
and C
60
calculated at B3LYP/6-31G(d) levelFig. 2
NMR spectra of B 3 N 3 C 54 and C 60 are calculated by gauge independent atomic orbital (GIAO) approach at B3LYP/6-31G(d) level. The NMR relative chemical shifts of nucleus ‘X’ are calculated by δ = δ ref − δ full , where δ full are isotropic magnetic shieldings of nuclei of fullerene and δ ref are those of reference systems which are C 4 H 12 Si for C, B 2 H 6 for B and NH 3 for N nucleus. The calculated chemical shift for C 60 is 131.8 ppm whereas that of B 3 N 3 C 54 lies between 121.5 and 143.9 ppm. The minimal δ value corresponds to C attached to C–B bond whereas maximal value belongs to C bonded to C–N moiety, as expected due to lone pair of N atom. The triply degenerate NMR peaks of B and N nuclei in B 3 N 3 C 54 are found to be at 10.1 and 162.8 ppm, respectively.
Electronic properties
The density of states (DOS) spectra along with molecular orbital energy levels of B
3
N
3
C
54
and C
60
are plotted in Fig.
3
. One can note that the energy levels of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of B
3
N
3
C
54
are slightly shifted upwards relative to C
60
. Furthermore, the substitution results in additional energy levels which are closely spaced and broaden the density of states spectrum. The DOS spectra also reveal that the HOMOs of C
60
are fivefold degenerate and LUMOs are triply degenerate. This degeneracy of molecular orbitals is lowered for B
3
N
3
C
54
in which the HOMOs and LUMOs are triply and doubly degenerate, respectively. The degenerate HOMOs and LUMOs of B
3
N
3
C
54
and C
60
are displayed in Fig.
4
. Within the framework of Koopmans’ theorem, the ionization potential (
I
) and electron affinity (
A
) are approximated by the negative energy eigenvalues of the HOMO and LUMO, respectively. From Table
2
, one can note that the calculated
I
and
A
values of B
3
N
3
C
54
are lower than those of C
60
. As known, it is energetically unfavorable to add electrons to a high-lying LUMO or to extract electrons from a low-lying HOMO and so to form the activated complexes of any potential reaction [
29
]. Therefore, B
3
N
3
C
54
is expected to be form cationic complexes due to smaller
E
HOMO
magnitude, i.e.,
I
value than that of C
60
.
Density of States (DOS) spectra of C
60
and B
3
N
3
C
54
calculated at B3LYP level. Occupied (
green
) and unoccupied (
blue
) molecular orbital energy levels are also displayed Degenerate HOMOs and LUMOs of B
3
N
3
C
54
(
upper set
) and C
60
(
lower set
) Electronic parameters of B
3
N
3
C
54
and C
60
at B3LYP/6-31G(d) level Parameter B3N3C54 C60 5.872 5.986 3.205 3.228 2.667 2.758Fig. 3
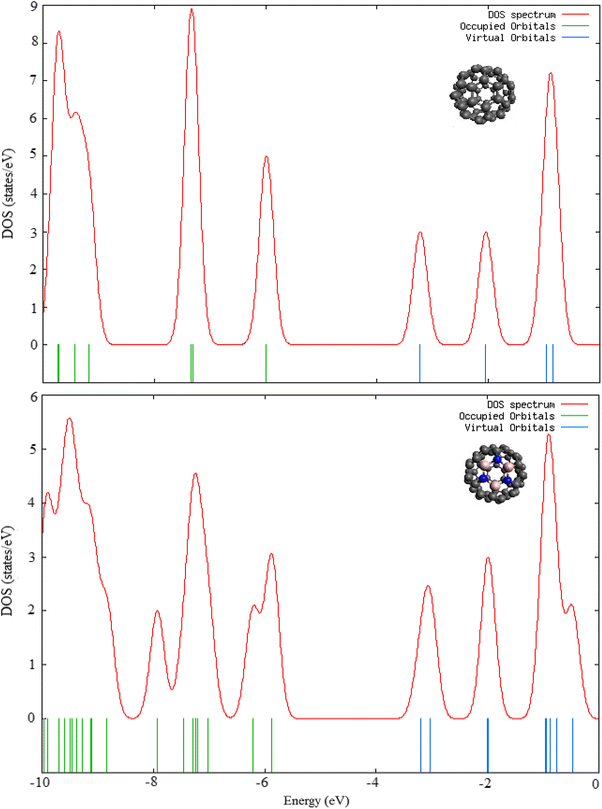
Fig. 4
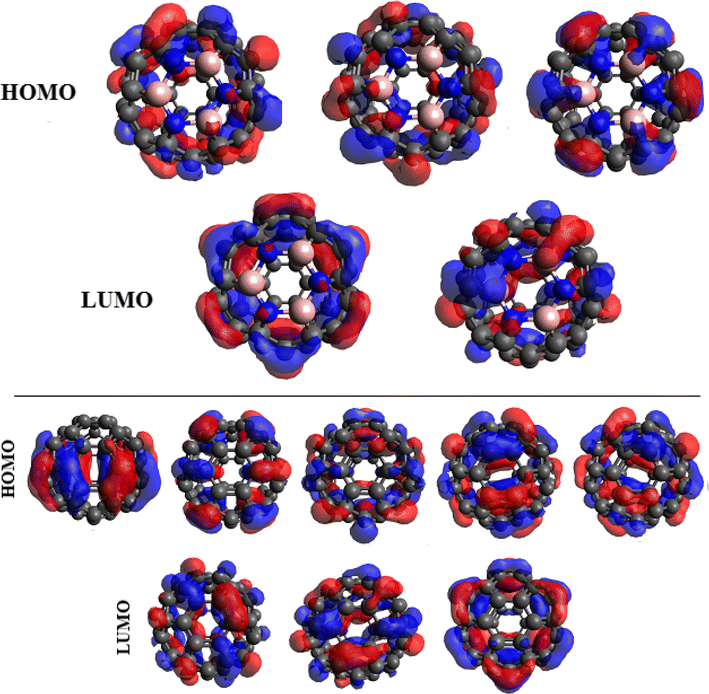
Table 2
The energy difference between HOMO and LUMO, HOMO–LUMO energy gap ( E gap ) corresponds to the band gap in solids which is equal to the difference of I and A values. In molecular system, this energy gap is generally approximated to the chemical hardness [ 30 ]. This energy gap is equivalent to the energy required to free an outer shell electron which behaves as a mobile charge carrier that moves freely within the solid material. The E gap is an important parameter that determines the electrical conductivity of a solid. Substances with larger energy gaps are generally insulators, materials with smaller energy gaps are semiconductors, and conducting materials have very small or no energy gaps. In other words, the molecules with smaller energy gaps are less hard, and hence more reactive. Our calculated E gap of C 60 at B3LYP/6-31G(d) level (2.76 eV) is in agreement with the experimental band gap value of 2.3 ± 0.1 eV [ 31 ]. This energy gap being neither too large nor too small suggests the semiconducting behavior of C 60 . In case of B 3 N 3 C 54 , this energy gap is reduced to 2.67 eV. Therefore, the conductivity of B 3 N 3 C 54 should be slightly greater than that of C 60 .