Graphical Abstract

Density functional theory and Time-dependent density functional theory (TDDFT)-based calculations were performed on surface-passivated Silicon nanocrystals (SPSNs) of different sizes. The surface passivation was achieved using H, F and Cl atoms. Various properties of the resulting optimized structures Si n H n , Si n H n−1 F and Si n H n−1 Cl ( n = 20, 24, 26 and 28) like binding Energy, dipole moment, HOMO–LUMO gap, vibrational IR spectra and absorption wavelengths were determined. Surface passivation studies reveal that all the SPSNs are insulators and TDDFT study performed using two basis sets 6-31G and 6-31+G (d,p) shows that the maximum optical absorption of all the samples lie in the UV region, except for Si 28 H 27 F SPSN which shows maximum absorption at ~588 nm. The absorption transitions in the Si n H n , Si n H n−1 F and Si n H n−1 Cl were characterized to be from pi→pi* transitions.
The field of research surrounding the optical and electrical properties of semiconductor nanocrystals has witnessed an enormous growth over the last decade [ 1 – 3 ]. Because of being compatible with conventional microelectronics and minimum feature size approaching to atomistic dimensions, silicon-based molecular electronics has emerged as a viable option [ 4 , 5 ]. Freestanding silicon nanocrystals hold great promise for a variety of applications such as photovoltaics [ 6 – 8 ], microelectronics [ 9 , 10 ], bioimagings [ 11 , 12 ], optoelectronics [ 13 , 14 ] and photosensitizing [ 15 , 16 ]. Furthermore, to design high density electronic devices for molecular computers, electronic properties of small organic molecules have been studied [ 17 , 18 ]; thus, extended studies for molecule–silicon junctions are also needed [ 19 – 21 ]. Especially, in an approach to molecular electronics the interconnection of organic molecules to semiconductors plays a vital role. Strongly doped silicon has been proved to be an excellent contact to interconnect to molecules, even more robust than gold [ 22 ]. Recent efforts are already oriented towards the characterization of hydrogen-passivated silicon interfaces for the synthesis of molecular electronic devices [ 23 ] as well as to study the effects produced by metals on organic molecules laying on hydrogen-passivated silicon.
Surface modification of silicon nanocrystals (Si NCs) is carried out to prevent them from oxidation [ 24 – 29 ]. Kauzlarich et al. [ 19 ] synthesized Cl-passivated Si NCs at room temperature in liquid phase and performed silanization, alkylation, and alkoxylation to modify them and found that the stability of Si NCs against oxidation dramatically increased after surface modification [ 30 – 32 ]. However, it was observed that no changes in photoluminescence (PL) from Si NCs were induced by surface modification [ 30 , 32 ]. However, in the work of Rogozhina et al. [ 35 ] the aminization of Cl-passivated Si NCs by the use of butylamine led to a redshift of the PL from Si NCs, which indicated that surface chemistry effect should be taken into account in the surface modification of Si NCs. Also, the sensitive dependence of the electrical, optical and magnetic properties of semiconductors on dopants has enabled an extensive range of semiconductor technologies [ 36 ]. Several new applications that require the discrete character of a single dopant, such as single spin devices in the area of quantum information or single-dopant transistors, demand a further focus on the properties of a specific dopant. It is well known that when the semiconductor technologies move into the nanometer-sized regime, dopants remain critical [ 37 , 38 ].
The size dependence of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) gap is one of the fundamental signatures of semiconductor nanocrystals or quantum dots, providing many unique physical properties and attracting considerable attention in optoelectronics, spintronics, photovoltaic, and biolabeling [ 39 – 45 ]. As the size of a semiconductor cluster shrinks, its optical gap and dipole oscillator strength increase with respect to the bulk value. Recently, many of the size-dependent optical properties of various semiconductor nanocrystals have been measured and characterized using several experimental methods [ 46 – 50 ]. Most of the theoretical effort has been devoted to calculations of the optical gap of semiconductor nanocrystals. However, the emission energy is actually most often measured and is usually more relevant for technological applications. In general, the absorption gap is larger than the emission gap; that is, light absorbed by semiconductor clusters is red-shifted to longer wavelengths on emission. This red shift is termed to as the Stokes shift, and its origin, magnitude, and size dependence have been a notable source of controversy in the recent literature, especially for SiNCs. Some measurements have found size-dependent Stokes shifts that decrease starting from 1.0 eV in small Si clusters [ 51 – 53 ]. Most recent work has demonstrated that single-walled carbon nanotubes when inserted into plants (termed as plant nanobionics) actually augment the photosynthesis and biochemical sensing [ 54 ].
In the present study, we have performed density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations on surface-passivated Silicon nanocrystals (SPSNs) using hydrogen, fluorine and chlorine atoms as surface passivants. All the SPSNs were then optimized for the lowest energy configurations. Several properties of the SPSNs, like binding energy, energy gap (HOMO–LUMO), dipole moment and absorption wavelengths were determined using the DFT and TDDFT calculations. To aid identification, infrared spectra have also been calculated.
The present calculations are performed using DFT and TDDFT. Self-consistent field (SCF) electronic structure calculations have been carried out on all the surface-passivated silicon nanocrystals (SPSNs), Si n H n , Si n H n−1 F and Si n H n−1 Cl with n = 20, 24, 26 and 28 (in total 12). Doped silicon-based nano-clusters of the same size happen to be promising nano-units in the emerging (low-dimensional) nano-opto-electronics [ 55 ]. The Pople′s N-31G split valence basis set 6-31G was used for all structures. Geometry optimizations were carried out using internal coordinates and with a convergence limit of 10 −7 hartree on the total energy. All the structures were built using the Avogadro package [ 56 ], and then the optimized structures were obtained by first relaxing the structures using the Steepest Descent algorithm in the Avogadro, without any symmetry constraints. It was followed by a more accurate calculation carried out using Becke’s three parameter functional with the Lee–Yang–Parr correlation functional (B3LYP) level of theory [ 57 , 58 ]. Similar DFT methods were successfully used to calculate the electronic properties of carbon clusters (fullerenes), similar in shape, symmetry and sizes [ 59 ]. Electronic properties were calculated from the SCF electronic energy and the orbital energy values. All theoretical calculations have been performed with the FIREFLY version 8.0.1 program code [ 60 , 61 ]. The optimized geometries were confirmed as minima on the potential energy surface (PES) by evaluating the hessian and checking for the presence of all real frequencies. The density of states plots was extracted from the DFT calculations with the help of Gausssum v3.0 package [ 62 ]. The electronic transitions between occupied and unoccupied states were calculated at Restricted Hartree–Fock (RHF) and TDDFT/B3LYP level of theory using basis sets 6-31G and 6-31+G (d,p), with 100 singlet excited states that result in the UV absorption spectra and the main orbitals contributing to the transitions. The optimized ground state configurations of all the SPSNs were used as starting configurations for the TDDFT calculations. TDDFT calculations were performed with no symmetry constraints. A very similar approach has been used in a recent TDDFT study on silicon-based clusters [ 63 ].
In the current study, silicon nanocrystals surface passivated with H, Cl and F atoms (Si
n
H
n
, Si
n
H
n−1
F and Si
n
H
n−1
Cl with
n
= 20, 24, 26 and 28) each have been considered. All the optimized structures (SPSNs) arranged categorically in the ascending order of their number of atoms can be seen in Fig.
1
.
Different Optimized SPSNs (
Red
Silicon,
Gray
Hydrogen,
Green
Chlorine,
Purple
Fluorine)Fig. 1

The binding energy is calculated using general Eq. (
1
):
The Binding Energy (in eV) plots shown in Fig.
2
display the stabilities of the SPSNs (Si
n
H
n
, Si
n
H
n−1
F and Si
n
H
n−1
Cl) with the increasing size
n
= 20, 24, 26 and 28. It has been consistently found that for all the SPSNs, the binding energy increases with the increasing value of ‘
n
’. It demonstrates that the formation of larger clusters is favored. In the present study, most favorable SPSNs are found to be Si
28
H
28
, Si
28
H
27
Cl and Si
28
H
27
F.
Variation of Binding Energy with the index
‘n’
of different SPSNsFig. 2

The HOMO–LUMO gaps/electronic gaps (in eV) were determined for all the SPSNs. As observed from the Fig.
3
, it can be noted the HOMO–LUMO gap varies between 4.57–4.96, 4.44–4.74 and 4.59–4.88 eV for Si
n
H
n
, Si
n
H
n−1
Cl and Si
n
H
n−1
F SPSNs. This band gap variation demonstrates the insulating nature of all the SPSNs, indicating the difficulty of transition of electrons from HOMO to LUMO orbitals. Additionally it can be extrapolated that the surface passivation with H, F and Cl atoms does not seem to play a major role in the conductivity of the SPSNs. It is to be noted that the HOMO–LUMO gaps are however underestimated in DFT calculations. In addition, it is to be noted that much smaller gaps (~1.5 eV) have recently been reported for hydrogenated silicon clusters of quite comparable size [
64
,
65
]. However, these small gaps resulted from the non-tetrahedral and over-coordinated structures of those clusters.
HOMO–LUMO Gap variation with the index
‘n’
of different SPSNsFig. 3

The density of states of all the SPSNs is shown in Fig.
4
. It is clearly seen that the spectrum of Si
24
H
23
Cl is the narrowest (between the virtual and occupied orbitals), while the spectrum of Si
26
H
26
is the widest, indicating that HOMO–LUMO gap of Si
24
H
23
Cl is smallest and that of Si
26
H
26
is highest.
Density of States of all SPSNs arranged in the order of increasing gap between virtual and occupied orbitals, from top to bottomFig. 4

The Fig.
5
shows variation of the dipole moment of all the SPSNs. The dipole moments were found to vary from 0.0 to 0.54, 2.5 to 3.73 and 2.0 to 2.73 Debye for Si
n
H
n
, Si
n
H
n−1
Cl and Si
n
H
n−1
F SPSNs, respectively. The maximum dipole moment/polarity has been found for the Si
28
H
28
, Si
24
H
23
Cl and Si
28
H
27
F in their categories, indicating the most polar nature. The Si
20
H
20
SPSN has been found to be non-polar.
Dipole moment variation with the index
‘n’
of different SPSNsFig. 5

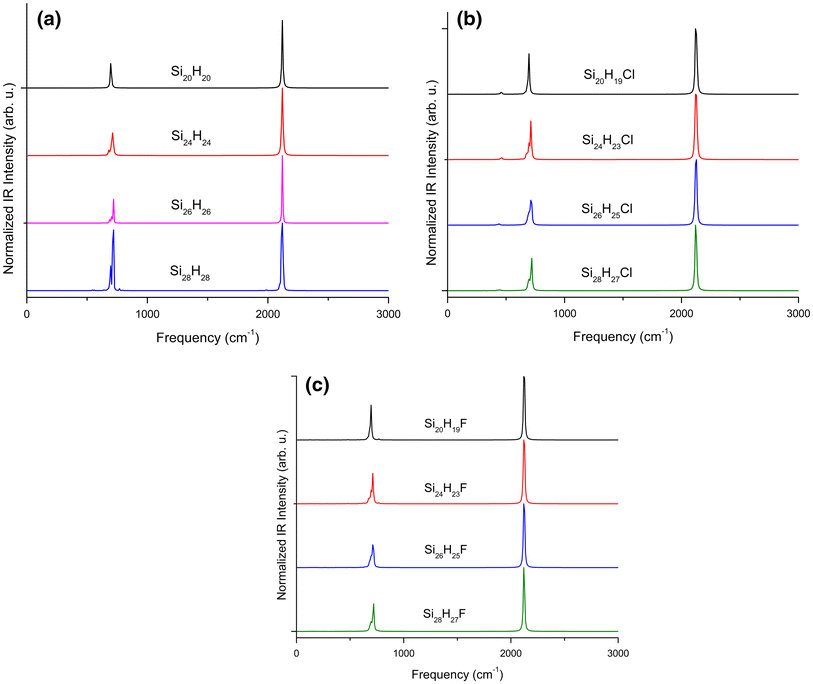
In this work, IR spectra have been calculated to validate the vibrational structures of Si
n
H
n
, Si
n
H
n−1
Cl and Si
n
H
n−1
F SPSNs, respectively. All the IR spectra can be seen in Fig.
6
, where the IR intensities are normalized. For all the SPSNs, Si–Si stretching fundamental frequencies were observed in the range 692–717 cm
−1
and Si–H stretching frequencies at 2121 cm
−1
(Fig.
6
a, b, c), which is in agreement with the work by Boo et al. [
66
]. In the Si
n
H
n−1
Cl SPSNs the Si–Cl fundamental frequency was observed in the range of 438– 461 cm
−1
(Fig.
6
b). For the Si
n
H
n−1
F SPSNs, we observed Si–H stretching fundamental frequency at 2121 cm
−1
which is same as that for Si
n
H
n
but Si–Si stretching fundamental frequency shifted in the range 700–716 cm
−1
. Unlike the work of Lucovsky et al. [
67
] who reported the Si–F stretching mode frequencies around 935 cm
−1
and Si–F bond bending frequencies below 400 cm
−1
, we observed weak shoulder peaks of Si–F bond with fundamental frequencies in the range 763–772 cm
−1
(Fig.
6
c). It is important to note from the figures that the intensities of Si–Cl and Si–F bonds are very less as compared to Si–H and Si–Si signature peaks, due to the fact that there are single Cl and F passivants atoms in all the SPSNs.
IR vibrational spectra of all the SPSNs indicating the signature peaks of Si–H, Si–Si, Si–F and Si–Cl bondsFig. 6
The maximum absorption wavelengths were also calculated for all the optimized SPSNs. TDDFT calculations show that all the SPSNs exhibit maximum UV absorption in the wavelength range 180–350 nm which constitutes the UV-B and UV-C regions. Table
1
comparatively lists the wavelength corresponding to the maximum absorbance (
λ
max
) and the maximum oscillator strengths, for two basis sets 6-31G and 6-31+G (d,p) and provides the molar absorption coefficients (
ε
max
) along with the major contributions with transition probabilities greater than 10 % for all the SPSNs, for larger basis set 6-31+G (d,p). It is clearly seen from the Table
1
that overall the
λ
max
values get red-shifted and oscillator strengths (
f
) have reduced/corrected with the larger basis set. The further discussion on optical absorption will be based on the 6-31+G (d,p) basis set. It is known that the oscillator strength is proportional to absorbance in a UV–Visible absorption spectrum. The
λ
max
values are found in the range 248.455–588.323 nm for all the SPSNs indicating a strong absorption capability of these SPSNs in ultraviolet region except for Si
28
H
27
F SPSN which has maximum absorption at 588.323 nm. In the case of Si
n
H
n
SPSNs, the maximum molar absorption coefficient (
ε
max
) is found for Si
24
H
24
(~6000 M
−1
cm
−1
) indicating the orbital transition to be from pi-bonding to pi-antibonding orbital transition (pi→pi*). In case of the Si
n
H
n−1
F SPSNs, pi→pi* transition is observed as the intense peaks with highest
ε
max
(~21000 M
−1
cm
−1
) for Si
28
H
27
F. For Si
n
H
n−1
Cl SPSNs, again the pi→pi* transition is assigned to the strong absorption with the highest
ε
max
(~6200 M
−1
cm
−1
) for Si
24
H
23
Cl. There was no consistent shift found in the
λ
max
found for all the SPSNs with increasing size. In all the SPSNs, lowest absorption wavelengths were found for the SPSNs with
n
= 20, and maximum absorption wavelengths were found for
n
= 24 SPSNs. It is also to be noted from Table
1
, that the major contributions to the transitions in all the SPSNs are from the deeper levels and not from the surface states (HOMO, LUMO). Moreover, several orbitals are playing role in the transitions. The highest contributions were found from H-11->L+2 (61 %) in case of Si
28
H
28
, H-10->L+2 (44 %) for Si
24
H
23
F and H-4->L+4 for Si
24
H
23
Cl.
TDDFT computed maximum absorption wavelengths (
λ
max
), oscillator strengths (
f
), molar absorption coefficients (
ε
max
) and orbital transitions (for 6-31+G (d,p) basis set) of all the SPSNs SPSN 6-31Ga 6-31+G (d,p)b Major contributions (>10 %) Si20H20 230.324 0.032 248.455 0.023 5000 H-12→L+2 (24 %), H-11→L+1 (14 %), H-9→L+3 (26 %) Si24H24 229.763 0.051 263.657 0.026 6000 H-10→L+1 (12 %), H-4→L+4 (25 %), H-3→L+5 (25 %) Si26H26 235.294 0.036 256.373 0.024 3400 H-8→L+3 (24 %), H-7→L+2 (24 %) Si28H28 236.799 0.009 257.046 0.010 4400 H-13→L+1 (20 %), H-11→L+2 (61 %) Si20H19Cl 228.001 0.030 250.881 0.014 5200 H-13→L+3 (16 %), H-12→L+3 (21 %), H-11→L+2 (20 %) Si24H23Cl 258.921 0.006 266.407 0.008 6200 H-9→L+2 (13 %), H-5→L+4 (12 %), H-4→L+4 (32 %) Si26H25Cl 234.488 0.016 258.005 0.007 3600 H-12→L+3 (12 %), H-8→L+3 (18 %), HOMO→L+11 (11 %) Si28H27Cl 266.431 0.009 259.074 0.007 3500 H-10→L+5 (13 %), H-9→L+6 (31 %) Si20H19F 232.286 0.015 249.935 0.014 5000 H-12→L+3 (32 %), H-11→L+2 (32 %) Si24H23F 242.368 0.013 261.989 0.015 5600 H-10→L+2 (44 %), H-4→L+5 (15 %) Si26H25F 232.547 0.019 257.919 0.007 3800 H-11→L+3 (32 %), H-8→L+3 (23 %) Si28H27F 562.323 0.098 588.218 0.071 21000 H-10→L+1 (24 %)Table 1
The binding energy variations show that all the SPSNs (Si n H n , Si n H n−1 F and Si n H n−1 Cl with n = 20, 24, 26 and 28) with higher sizes (corresponding to ‘ n ’) are most favorable. HOMO–LUMO Gap calculations show that all the SPSNs demonstrate an insulating nature. Slight variations in the band gaps with lowest gap (4.44 eV) for the Si 24 H 23 Cl SPSN and highest for Si 26 H 26 (4.96 eV) were found. This could indicate that there is no significant effect of surface passivation or even the cluster size on the conductivity of all the SPSNs.
The maximum absorption wavelengths were obtained from TDDFT calculations. The maximum wavelength absorption for all the SPSNs was found to lie in the UV region except for Si 28 H 27 F SPSN which has maximum absorption at 588.323 nm. The absorption transitions in the Si n H n , Si n H n−1 F and Si n H n−1 Cl were characterized to be from pi→pi* transitions. All the SPSNs studied in the present study could be effectively used in the applications of optical filters, photodetectors and plant nanobionics (to enhance the solar energy capture).
One of the authors (SCH) acknowledges the National PARAM Supercomputing Facility (NPSF) of C-DAC, Pune, India for providing the cluster computing facility.