Nuclear Research Center at Draria (CRND), Algiers, DZ
M’Hamed Bouguerra University, Boumerdes, DZ
Abstract
In the present work, adsorption and dif fusion of oxygen (O) atom on uranium dinitride (UN
2
) is studied to map out the preferential UN
2
(100) surface site. The first principle method based on density functional theory (DFT) within the generalized gradient approximation PBE and the covariant version energy functional PBE + U correction were used. The supercell approach and a coverage dependence of the adsorption structures and energetic were studied in detail for several monolayers’ (ML) range. Potential energy surfaces (PES) corresponding to the interaction between O atom and UN
2
(100) on surface and subsurface for several sites and layers (Top U and Top N slabs) were calculated and favorable sites were identified with their maxima energy stable positions, which were then analyzed. For all positions, the PES show the same system behavior, when the O atom is sufficiently far from the UN
2
surface, and the energy of the system tends to the sum of free UN
2
slab and free oxygen atom energies. In return, when the distances decrease, strong interactions appear with presence of important potential wells. Calculation results showed that favored on-surface site for O atom adsorption were found to be near the bridge one for the UN (Top U slab) corresponding to five layers, uranium terminated and top one for (Top N slab) corresponding to six layers nitrogen terminated, the maximum system energy is situated at a position of about 1.2 and 1.5 Å from the surface for the two layers types calculations respectively. For subsurface results, only Top N presents a favorable incorporation site at the hollow position and the penetration of O atom is about −0.5 Å from the surface. DFT + U study confirms all the results obtained by DFT calculations; that is, the maxima site positions for oxygen atom and the adhesion energy values per atom are of the same order of magnitudes. The adsorption energy per oxygen atom and the mean distance from the top surface gradually decrease with the coverage of O atoms for both on-surface cases, Top U and Top N slabs, with oxygen occupying the favorable site. For the Top N slab hollow site, the incorporation of oxygen through the surface becomes effective from a coverage of 3/8 ML with an encrustation of about −0.3 Å.
Introduction
Nowadays, uranium nitrides constitute an alternative advanced nuclear fuel that has great prospects to replace actual oxide fuels. This interest is due to some very interesting physical properties: high thermal conductivity, very high melting point, and high density which are very appropriate for future fast breeder and high-temperature reactors (small core with high power density…).
Uranium nitride refers to a family of several ceramic compounds: uranium mononitride (UN), uranium sesquinitride (U
2
N
3
), which exists in either an alpha or beta phase, and uranium dinitride (UN
2
).
The oxidation of uranium mononitride in an oxygen atmosphere was systematically studied both experimentally and theoretically, as mentioned in many references [
1
–
3
]. It was demonstrated that oxygen impurities’ incorporation even in little quantities into this fuel alters its physical properties (thermal conductivity,…); however for dinitrides and sesquinitrides, only some efforts have been devoted to these types of fuel. Some recent experimental works (powder XRD analysis) have revealed that up to 1.0 wt% uranium oxides (UO
2
) were identified in UN
2
and U
2
N
3
[
4
].
In this work, adsorption of oxygen adatom on UN
2
surface is studied to understand this phenomenon thoroughly. First principle study is used to follow how oxygen atom is adsorbed or incorporated on/into the (001) surface by calculating relative adsorption energies of oxygen atoms on different UN
2
(100) sites and by determining the potential energy surface (PES) of the system.
Several sites were chosen by considering the symmetry of the UN
2
(100) surface system, and two types of slabs were selected considering surface and work function calculus variation. The first one is the five (5)-layer uranium-terminated slab (Top U) and the second one consists of the six-layer-terminated nitrogen (Top N) slab. When the favorite sites (on/in UN
2
(100) surface) that are determined, we used the concept of DFT + U exchange correlation developed by Dudarev et al. [
5
] to confirm these maxima positions, since uranium belongs to the strongly correlated materials family, which is characterized by strong Coulomb correlation among the partially filled 5f electrons that cause unreasonable ground state properties [
6
].
We also performed calculations of coverage ordered structures at the identified preferential adsorption sites, with (1 × 1), (2 × 1) and (2 × 2) slabs of supercells corresponding to different mono-layer values depending on the Top U or Top N slab surface and the preferential site study. For every coverage we performed a full structure optimization, including substrate relaxations. Of particular interest was to determine oxygen atom adsorption, energy progression and the mean oxygen distance to the top surface.
Calculation method and definitions
The calculations were performed using the DFT total energy calculation carried out by Vienna ab initio simulation package [
7
,
8
]. We employed the projector augmented wave method [
9
] (PAW) and the generalized gradient approximation (GGA) [
10
] based on Perdew–Burke–Ernzrhof (PBE.52) for the exchange correlation functional. The wave functions are expanded in a plane-wave basis set with an energy cutoff of 550 eV. The surface is modeled by a Top U five-layer slab and a Top N six-layer slab, separated by 20 Å of vacuum space. Oxygen is placed on one side of the slab where the induced dipole moment is taken into account by applying a dipole correction [
11
]. We allow atomic relaxation of all atoms in the top three layers (Top U) and four layers (Top N) of the slabs and the oxygen atoms falling down to the surface. The final forces on the atoms are less than 0.01 eV/Å. For the k-point sampling, a 5 × 5 × 1 mesh of Γ centered points is used for the 001 surface unit cell [
12
].
The adsorption energy (
Eadsorption
) is always considered as a measure of the strength of adsorbate–substrate adsorption. It is defined as [
13
,
14
]:
where
Eadsorption
is the adsorption energy,
Eadsorbate_substrate
the total energy of the adsorbate–substrate system in the equilibrium state,
Esubstrate
the total energy of the substrate alone, and
Eadsorbate
the total energy of the free adsorbate alone.
With this definition, positive values of
Eadsorption
reflect strong interaction of the adsorbed species with the surface atoms. The average adsorption energy per oxygen atom on the surface,
Ead_O
, is defined as:
where
NO
is the number of oxygen atoms in the surface unit cell, and
EO/UN2
,
EUN2
, and
EO
represent the total energy of the adsorbate–substrate system, the clean surface, and the free oxygen atom, respectively. So, a positive number indicates that the adsorption is exothermic (stable) and a negative number indicates that it is endothermic.
The adsorption energy per oxygen atom can also be referenced to the energy which the O atom has in the O
2
molecule by subtracting half the binding energy
,
Atomic oxygen adsorption on UN2(100) surface
The adsorption of atomic O on UN
2
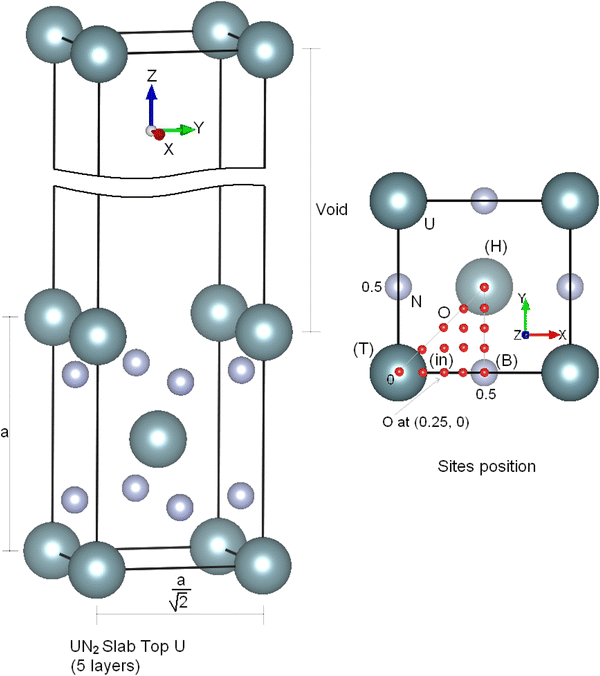
(100) surface is calculated with one atomic oxygen in every (1 × 1) surface unit cell. Several distinct adsorption sites in agreement with the symmetry slab are examined. These alternative adsorption sites are mentioned in Fig.
1
as (H): hollow, (B): bridge, (T): top, (H-B): hollow-bridge, (H-T): hollow-top, (T-B): tTop-bridge and (in): site between (H), (B), and (T) and so on. We adopt these coordinate references for the Top N study it is clear that bridge site (0.5,0) for five slab U terminated correspond in reality to a top site with the same (0.5,0) coordinate for six slab N terminated (see Fig.
1
).
Fig. 1
UN
2
Slab and adsorption sites
Adsorption energies were calculated at different positions of every site to find out which one of them was the most favorable for oxygen adsorption in and on the UN
2
(100) surface.
By varying oxygen atom positions in the
z
axis, a curve representing adhesion energy versus
z
position (distance from surface) is determined for every site and then, different maxima are deducted (Top U and Top N separately).
The bulk UN2, UN2(100) surface and oxygen
Bulk calculation was performed without spin polarization consideration, since free UN
2
is a nonmagnetic (NM) phase through PBE52 calculation [
15
]. The calculated lattice constant of UN
2
is 5.276 Å (experimental value 5.31 [
16
]) and the bulk modulus calculated by using the third-order Birch–Murnaghan equation of states (EOS) [
17
] is 250 GPA in the order of other theoretical values (252 [
15
], from 235.8 to 264.6 Gpa [
18
]). Nevertheless, the lattice constant was somewhat smaller than the experimental value.
A DFT + U formalism developed by Dudarev et al. [
5
] which consists of adding a depending functional on the parameter U to the conventional one to force the on-site Coulomb repulsion in the uranium atom [
19
] was used to correct the lattice parameter. To achieve the experimental lattice parameter value (5.31 Å), the optimal Hubbard
U
was found equal to
Uoptim
= 2.6 eV which assures a new bulk modulus of 251 GPA.
Since the triplet state is the ground state of the free O [
13
,
20
], all our surface DFT calculations were performed using spin polarization method with DFT and DFT + U theories.
The variation of the work function and surface energies remains roughly constant by varying layers number of the slab (
W
≈ 2.9 eV;
ESurf
≈ 5.2 J/m
2
for Top U slabs: 5 layers, 7, 9 and 11; similarly,
W
≈ 7.2 eV;
ESurf
≈ 6.2 J/m
2
for Top N slabs: 6 layers, 8, 10 and 12 surface cells); our study concerns five layers for Top U and six layers for Top N.
The binding energy of O
2
,
, is 6.49 eV from DFT-PBE52 ion relaxation calculation and the bond length is
dO–O
= 1.23 Å. The experimental values are
= 5.12 eV and
dO–O
= 1.21 Å [
21
]; the binding energy is therefore overestimated and is well known for ab initio theoretical results (
= 6.67, 6.24 [
10
,
22
]). DFT free oxygen calculation is not affected by the DFT + U method, since no
f
or
d
valence electrons belong to oxygen atom.
Results and discussion
Atomic oxygen adsorption on UN2(100) surface spin and no spin consideration
We have performed some atomic oxygen adsorption on UN
2
surface by considering spin and no spin configurations. Figure
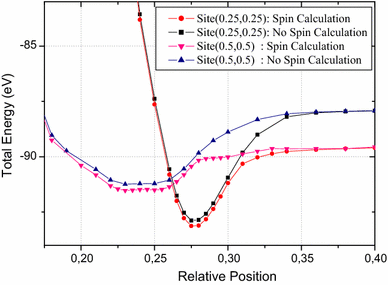
2
shows Top N calculations for two sites (0.25,0.25) and hollow (0.5,0.5) ones.
Fig. 2
PES oxygen adatom total energy comparison for hollow site and (0.25,0.25) one on UN
2
(001). Top N surface of calculation with and without spin polarization
From Fig.
2
, one can observe the difference between the spin and no spin calculation, i.e., far from the surface the total energy is equal to the sum of free oxygen and free UN
2
total energy, and when oxygen is close up to the surface the interaction becomes effective and the deviation of the two calculations is reduced. Nevertheless in this work, all minimal total energies were carried by the spin calculations.
UN2 Top U slab results
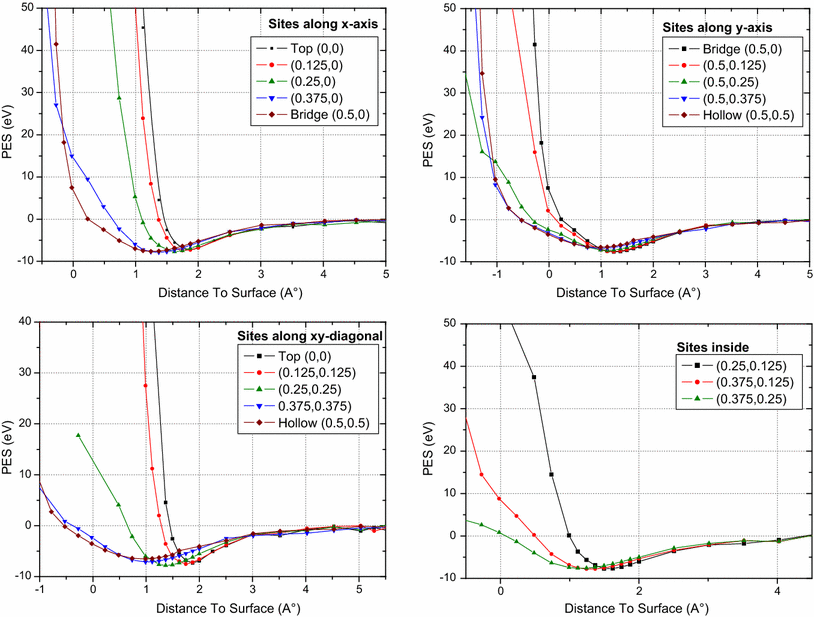
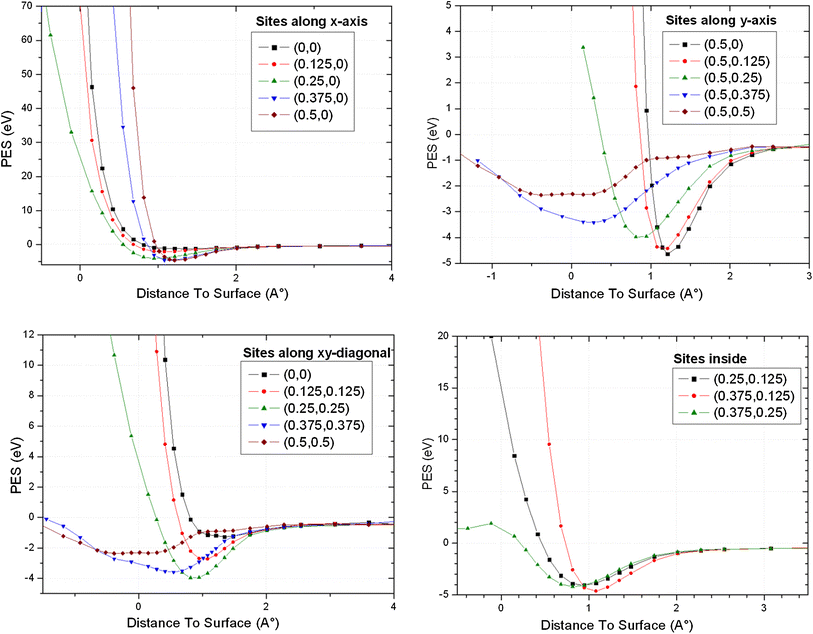
Results of oxygen atom approaching UN
2
Top U surface for several sites are indicated in Fig.
3
. All the results show minima total energy on the top surface at a distance situated between 1 and 2 Å, constituting favorable adsorption positions; however, the favorable site corresponding to greater energy adsorption is situated at (0.375,0) near the bridge site, in which the energy results are of the same magnitude.
Fig. 3
PES oxygen adatom for several sites along the
z
axis for Top U UN
2
surface
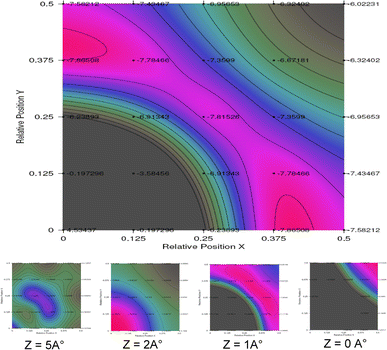
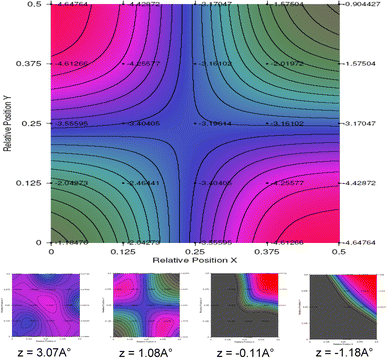
Interface plots in Fig.
4
show PES allure (
x
,
y
) at different
z
positions (height from surface), principal one in the figure corresponds to the most probable adsorption position
z
= 1.17 Å, favorable site adsorption is clearly established around the bridge (0.5,0) site (see red color).
The other slight figures show PES allure at different distances from the UN
2
(100) surface. At 5 Å height, the probability of oxygen adsorption is practically the same; then, at 2 Å height, the preference go for the top (0,0) site.
At the region of 1 Å, the bridge (0.5,0) site constitutes the more favorable one, then the probability of adsorption shifts to the hollow (0.5,0.5) site at a height of 0 Å (just on the surface, no material obstacle, no U or N atoms).
Fig. 4
Variation of interface energy at different levels from UN
2
Top U surface (
z
= 1.17 Å = principal) as a function of lateral displacement of oxygen atom
UN2 Top N slab results
For the case of UN
2
Top N surface approached by oxygen atom at different sites, one can see from Fig.
5
the same phenomenon of high probability of adsorption at a distance of about 1 Å from the surface for most of the position sites studied; nevertheless, the top site (0.5,0) is clearly the most favorable on site at
z
= 1.2 Å (see Table
1
). We remark for this case that the sites next to the hollow one began to present diffusion through the top surface and became effective for the hollow site with an incorporation of about −0.5 Å.
Fig. 5
PES oxygen adatom for several sites along the
z
axis for Top N UN
2
surface
Table 1
Maxima position for Top U and Top N layers (DFT, no relaxation calculation)
Site (position)
PES O atom (eV)
Position/surface (Å)
Top U on surface
(0.375,0)
−7.86
1.17
Top U on surface
(0.5,0) bridge
−7.65
1.17
Top N on surface
(0.5,0)
−4.65
1.21
Top U subsurface
None
–
–
Top N subsurface
(0.5,0.5) hollow
−2.32
−0.38
Interface plots confirm these results in Fig.
6
, where it is noticeably clear that the on the top surface the favorable site is situated at the (0.5,0) site, and the favorable incorporation site is the hollow (0.5,0.5) one.
Fig. 6
Variation of interface energy at different levels from UN
2
Top U surface (
z
= 1.2 Å = principal) as a function of lateral displacement of oxygen atom
UN2 Top U and Top N slabs’ energy adsorption
We report in Table
1
maxima energies results taken from Figs.
3
and
5
, with positions, energy and distances from surface obtained by PES method. To confirm these locations data, an ion relaxation calculation is necessary (see below).
Adsorption energies were then calculated for the most favorable site obtained by DFT calculation; DFT + U method was also used for a comparison study.
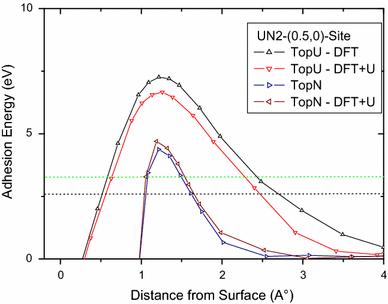
We remark from Fig.
7
that the shape of the on-surface curve results are similar for the two calculation types, DFT and DFT + U, in that on-surface energy oxygen adsorption are at the same order of magnitude for the Top U and Top N cases and maxima positions are practically at the same distance from the surfaces. For the maxima positions, adsorption is exothermic (stable).
Fig. 7
Comparison of DFT and DFT + U calculation for oxygen energy adsorption on UN
2
(100) surface through the (0.5,0) site
The dotted line in the curve indicates the limit of O adsorption stability if the adsorption energy is considered with respect to half the binding energy (see Eq.
2
) of theoretical O
2
molecule (green dot line) and experimental O
2
molecule (black dot line). For the maxima positions, adsorption remains exothermic (stable).
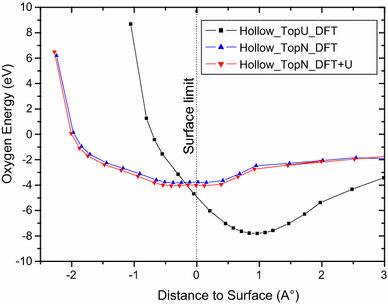
From Fig.
8
one can see the same remarks for the hollow site where the O energies are the same order of magnitude for DFT and DFT + U calculations and the oxygen incorporation is practically at the same distance. It is noticed from Fig.
8
that incorporation from the hollow site Top U surface is not allowed.
Fig. 8
Comparison of DFT and DFT + U calculation for PES in surface oxygen incorporation
As mentioned above, maxima positions were recalculated using DFT and DFT + U technique with the ion relaxation calculation. The positions are situated on (0.5,0) site for oxygen on-surface adhesion (Top U and Top N slabs) and in-surface Top N slab hollow (0.5,0.5) site O incorporation.
From Table
2
, the on-surface Top U preferential bridge oxygen adsorption is confirmed at around 1.13 Å for DFT and DFT + U results; the oxygen energy adsorption is at the same order of magnitude, 7.795, for DFT and 7.283 for DFT + U results. The magnetizations are practically the same.
Table 2
Maxima position (DFT and DFT + U ion relaxation calculation)
UN2(100) surface site
Calculation method
Adhesion energy of oxygen atom (eV)
Mean distance of oxygen atom to surface (Å)
Magnetization (μB)
Top U-bridge (0.5,0)
DFT
7.795
1.134
3.849
Top U-bridge
DFT + U
7.283
1.139
3.842
Top N (0.5,0)
DFT
6.139
1.539
3.762
Top N (0.5,0)
DFT + U
6.257
1.537
3.934
Top N-hollow (0.5,0.5)
DFT
3.573
−0.481
2.246
Top N-hollow
DFT + U
3.618
−0.492
2.087
For on-surface Top N slab, all the results are at the same magnitude for the DFT and DFT + U methods. It is noticed that the mean bridge position of oxygen is passed to 1.53 Å from the surface and the two nitrogen atoms of the top surface (supercell 1 × 1) show a little deviation in the
z
axis (about 0.7 Å).
For in-surface Top N slab UN
2
(100) surface, the hollow site is confirmed and the preferential position is at ~−0.5 Å through the top surface with a binding energy of around 3.6 eV (both DFT and DFT + U method).
Coverage study
Adsorption of atomic oxygen at different coverages on the UN
2
(100) surface is examined using density functional theory (DFT) and the Hubbard correction DFT + U formalism to investigate the variation of monolayer (ML) effects on the surface properties, in particular the adhesion energy per O atom and the oxygen mean distance to the surface.
Surfaces studied are (1 × 1), (2 × 1) and (2 × 2) supercells, where oxygen atoms occupy bridge sites for on-surface Top U, which corresponds to coverage from 1/4 to 2 monolayer, and Top N, which corresponds to coverage from 1/8 to 1 monolayer.
For the in-surface Top N surface case, oxygen atoms occupy hollow sites and the coverage values go from 1/8 to 1/2 monolayer.
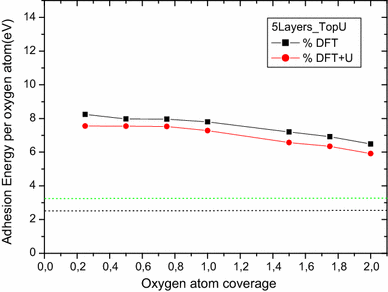
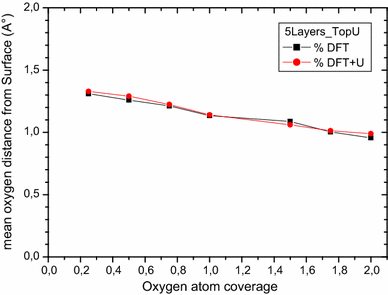
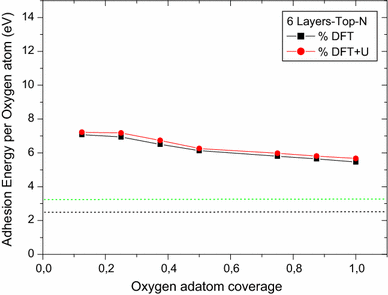
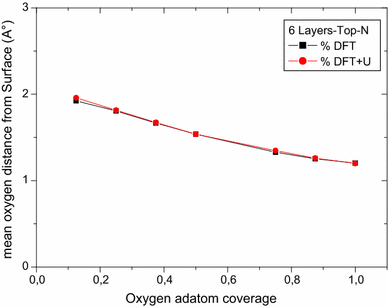
For on-surface oxygen atom adhesion, both Top U and Top N cases show an oxygen adsorption energy per atom and the mean distance from the surface decrease as the coverage increases. Both DFT and DFT + U calculations give the same profile of data and results are at the same order of magnitude (see Figs.
9
,
10
,
11
,
12
). Nevertheless, Top U study shows more deviation results (less 10 %) on comparing DFT and DFT + U calculations. It is also noticed that Top U free supercell calculations (DFT or DFT + U) provide magnetization results more superior than the Top N ones (~5.7 μB for Top U and ~2 μB for Top N).
Fig. 9
Adhesion oxygen energy per atom vs coverage monolayer for UN
2
(100) Top U surface slab
Fig. 10
Mean oxygen distance from surface vs coverage monolayer for UN
2
(100) Top U surface slab
Fig. 11
Adhesion oxygen energy per atom vs coverage monolayer for UN
2
(100) Top N surface slab
Fig. 12
Mean oxygen distance from surface vs coverage monolayer for UN
2
(100) Top N surface slab
The dotted lines in the curve (Figs.
9
and
11
) specify the limit of O adsorption stability if the adsorption energy is considered with respect to half the binding energy of theoretical O
2
molecule and experimental one. Calculations predict that for all coverage studied (both Top U and Top N with DFT and DFT + U) the O adsorption is stable.
For the encrustation of oxygen through the Top N slab at the hollow site versus O coverage, we found that below 3/8 ML, oxygen atom is ejected out of the surface (for ML = 1/8 and 1/4 the oxygen is at near +0.2 Å on the surface) with an adhesion energy per O atom of 1.8 and 2.9 eV, respectively. By increasing O atom number, the incorporation becomes effective (for ML = 3/8, 1/2, the mean distance oxygen–surface is equal to −0.3 and −0.5 Å, respectively, with an incorporation energy of about 3.5 and 3.6 eV, respectively) for both DFT and DFT + U studies.
Conclusion
Atomic oxygen adsorption on uranium dinitride, UN
2
(001), surface was studied by performing ab initio total energy and electronic structure calculations based on the density functional theory (DFT) and the covariant version of the DFT + U energy functional proposed by Dudarev et al. Several sites of UN
2
(100) surface and two kinds of surface slabs (5-layer Top U and 6-layer Top N) were considered in this study.
Calculation results (DFT and DFT + U) showed that the oxygen atom preferentially adsorbs on the bridge sites for the Top U and top site (0.5,0) in Top N slabs surfaces; nevertheless, only Top N slab permits incorporation of oxygen atom in the hollow (0.5,0.5) site.
The favored positions of oxygen atoms at the (0.5,0) site for Top U and Top N slabs in relation to top surface layers are at ~1.13 and ~1.53 Å, respectively, with an adhesion energy per atom of ~7.8 and ~7.2 eV, respectively, for DFT and DFT + U methods of calculation. The more favorable position in the hollow site for the Top N slab is situated at −0.48 Å (confirmed by DFT + U −0.49 Å) with 3.6 eV of oxygen atom incorporation energy for both calculations.
For coverage study of both DFT and DFT + U, the adsorption energy per oxygen atom and the mean distance from the top surface gradually decreased with the coverage of O atom for both on-surface cases, Top U and Top N, with oxygen-occupying bridge site. The calculations predict that for all coverage studied (both Top U and Top N with DFT and DFT + U) O adsorption is stable. Diffusion through the surface at the hollow site for the Top N slab becomes effective from the coverage of 3/8 ML.
All favorable sites remained stable with the adsorption energies diminished by a value of half the binding energy of oxygen,
.
Arai et al. (1993) The effect of oxygen impurity on the characteristics of uranium and uranium–plutonium mixed nitride fuels (pp. 70-78) 10.1016/0022-3115(93)90030-3
Sunder and Miller (1998) XPS and XRD studies of corrosion of uranium nitride by water (pp. 568-572) 10.1016/S0925-8388(98)00157-1
Chinthaka-Silva et al. (2008) Microscopic characterization of uranium nitrides synthesized by oxidative ammonolysis of uranium tetrafluoride (pp. 3076-3084) 10.1021/cm7033646
Dudarev et al. (2000) Understanding STM images and EELS spectra of oxides with strongly correlated electrons: a comparison of nickel and uranium oxides10.1016/S0968-4328(99)00115-8
Anisimov et al. (1993) Density-functional theory and NiO photoemission spectra10.1103/PhysRevB.48.16929
Kresse and Furthmuller (1996) Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set10.1016/0927-0256(96)00008-0
Kresse and Furthmuller (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set10.1103/PhysRevB.54.11169
Neugebauer and Scheffler (1992) Adsorbate–substrate and adsorbate–adsorbate interactions of Na and K adlayers on Al(111) (pp. 16067-16080) 10.1103/PhysRevB.46.16067
Monkhorst and Pack (1976) Special points for Brillouin-zone integrations10.1103/PhysRevB.13.5188
Zhang et al. (2011) Adsorption and dissociation of O2 on CuCl(1 1 1) surface: a density functional theory study (pp. 408-413) 10.1016/j.apsusc.2011.08.130
Shi and Stampfl (2007) First-principles investigations of the structure and stability of oxygen adsorption and surface oxide formation at Au(111)10.1103/PhysRevB.76.075327
Lu et al. (2011) Structural, electronic, mechanical, and thermodynamic properties of UN2: systematic density functional calculations (pp. 46-51) 10.1016/j.jnucmat.2010.12.308
Rundle et al. (1948) The structures of the carbides, nitrides and oxides of uranium10.1021/ja01181a029
Kotomin et al. (2007) Atomic scale DFT simulations of point defects in uranium nitride
Dudarev et al. (1997) Effect of Mott–Hubbard correlations on the electronic structural stability of uranium dioxide10.1080/13642819708202343
Zhang et al. (2011) Adsorption and dissociation of O2 on the Cu2O(1 1 1) surface: thermochemistry, reaction barrier (pp. 4787-4794) 10.1016/j.apsusc.2010.12.040
Nagai et al. (2005) Kinetic study of the quenching reaction of singlet oxygen by flavonoids in ethanol solution 109(9) (pp. 4234-4240) 10.1021/jp0451389
Heng-Rui et al. (2011) First principles study of adsorption of O2 on Al surface with hybrid functionals10.1063/1.3665032