Graphical abstract

The chemistry of hoop-shaped π-conjugated molecules has increased dramatically in recent years. We present here a computational modeling of photoinduced electron transfer processes in a series of host–guest complexes of Twin1 , Twin2 , and Twin3 double nanohoops with C 60 fullerene. According to our findings, charge transfer from cycloparaphenylene (CPP) fragments to C 60 is energetically favorable and occurs on a sub-nanosecond time scale. The slow decay of the generated charge-separated state suggests that the complexes may be of interest for organic photovoltaics.
Molecular systems with unusual topology attract significant attention from scientists of different research fields. Their electronic and chemical properties, as well as esthetic beauty, encourage scientists to look for new distorted, strained, bend or interlocked molecules [ 1 , 2 , 3 – 4 ]. Highly expanded π-conjugated architectures hitherto unknown are of great interest for photovoltaic applications. An example of such systems with unusual topology are cycloparaphenylenes (CPPs)—radially π-conjugated nanohoops constructed of para -linked phenylene rings [ 5 ]. Over the past 10 years, significant developments in organic synthesis have allowed for the precise control of the amount of phenylene units in the target nanohoops, resulting in CPPs with a diameter range of 7–28 Å. The variability in the size of nanohoops has led to their wide application in supramolecular chemistry [ 5 , 6 ]. A change in size dramatically affects their photophysical properties [ 7 , 8 – 9 ]. The first host–guest complex of CPP was reported by Iwamoto et al . in 2011 [ 10 ]. The authors found that [10]CPP with ten phenylene units has a nearly ideal diameter (13.8 Å) to accommodate C 60 fullerene. Bigger CPPs can bind larger fullerenes and endohedral metallofullerenes [ 11 , 12 – 13 ]. The formation of fullerene-nanohoop host–guest complexes suppresses fullerene self-aggregation, which encourages their use as a component of organic solar cells. It has been reported that certain supramolecular donor–acceptor (DA) complexes of CPPs with different fullerenes are capable of photoinduced electron transfer (PET). CPPs and their π-extended analogs serve as an electron-donating species in many of them [ 14 , 15 ].
In 2021, Du and co-workers [
16
] presented an unusual double-nanohoop molecule (
Twin1
)—a highly strained all-phenylene bismacrocycle, termed conjoined (1,4)[10]cycloparaphenylenophane. It consists of two CPP units linked by a twisted benzene ring (Fig.
1
). The absorption spectrum of
Twin1
is similar to that of [10]CPP, while the extinction coefficient is twice as high as that of the parental CPP. However, the emission spectrum of
Twin1
demonstrates a significant redshift (of more than 50 nm) compared to [10]CPP. The authors presume that the observed shift is associated with a high strain energy (greater than 110 kcal/mol), and the corresponding enhancement of vibrational couplings. The
Twin1
bismacrocycle exhibits a suitable diameter to form inclusion complexes with
C
60
. The binding constant was determined to be
K
a1
= (7.46 ± 0.33) × 10
5
M
−1
and
K
a2
= (5.85 ± 0.25) × 10
4
M
−1
for the mono (
Twin1 ⊃ C
60
) and the bis (
Twin1 ⊃ 2C
60
) adduct, respectively.
Structure of the double-nanohoop systems studied in this workFig. 1

Also, Juríček and co-workers [ 17 , 18 ] reported a new member of the double-nanohoop family ( Twin2 )—a giant π-conjugated framework with two CPP units linked by a peropyrene fragment (Fig. 1 ). The absorption spectrum of Twin2 was found to be a superposition of absorption spectra of CPPs and peropyrene, while the emission spectrum is nearly identical to that of the peropyrene unit. The authors demonstrated that such a system, with the bay region of central peropyrene core embedded in nanohoops, can form stable complexes with C 60 fullerene. They managed to obtain Twin2 ⊃ C 60 complex, while a bis adduct has never been observed in the solid state.
In 2022, Zhu, Cong, and co-workers [ 19 ] successfully synthesized a double nanohoop ( Twin3 ), which combines two [10]CPP units and a flexible cyclooctatetrathiophene core. This combination was found to be essential for the formation of host–guest complexes with C 60 and C 70 fullerenes. The binding constant for the mono ( Twin3 ⊃ C 60 ) and the bis ( Twin3 ⊃ 2C 60 ) adducts was found to be K a1 = (1.3 ± 0.1) × 10 5 M −1 and K a2 = (1.9 ± 0.2) × 10 4 M −1 , respectively.
In this paper, we present a computational investigation of the structural, electronic, and photoinduced electron transfer properties of host–guest complexes of the Twin1 , Twin2 , and Twin3 double nanohoops with C 60 fullerene. We compare the rates of the formation and the decay of charge transfer (CT) states to find the best candidate for further experimental research. Our results shed light on the role of structure and electronic nature of the linker between two CPP units in electron transfer processes.
Geometry optimizations were performed using ORCA 4.2.1 program [ 20 ]. BLYP [ 21 , 22 ] functional was used with def2-SVP basis set [ 23 , 24 ]. Resolution of identity approximation [ 25 , 26 ] and D3 dispersion correction by Grimme with Becke-Johnson damping [ 27 , 28 ] were also employed. Vibrational frequencies were computed for all structures. The absence of imaginary frequencies confirmed that the obtained geometries correspond to the minima on the potential energy surface. The interaction energy of the complexes was computed at the BLYP-D3(BJ)/def2-TZVP level of theory. BLYP-D3(BJ) functional was chosen as the DFT functional with the best accuracy-to-cost ratio for non-covalent interactions [ 29 , 30 ]. As previously demonstrated, using range-separated functionals is required for accurate prediction of charge transfer rates [ 31 , 32 – 33 ]. Consequently, vertical excitation energies were calculated using Tamm–Dankov approximation (TDA) formalism [ 34 ] with range-separated CAM-B3LYP [ 35 ] functional and def2-SVP basis set, [ 22 , 23 ] using Gaussian 16 (rev. A03) [ 36 ]. The same program was used for population analysis and calculation of Mulliken [ 37 , 38 ], Lowdin [ 39 ], Hirshfeld, [ 40 ] and CM5 [ 41 ] charges. Energy decomposition analysis (EDA) was performed using the Amsterdam Density Functional (ADF) program [ 42 ]. Topological analysis of the electron density distribution was conducted using the “Quantum Theory of Atoms in Molecules” (QTAIM). [ 43 ] AIMALL package [ 44 ] was applied to evaluate the bond critical point properties and associated bond descriptors. Excited states were analyzed by constructing natural transition orbitals (NTO) introduced by Luzanov et al . [ 45 ] and implemented within modern many-body codes by Head-Gordon et al . [ 46 ] Chemcraft 1.8 [ 47 ] was used to visualize chemical structures and frontier molecular orbitals.
Interaction energy, Δ
E
int
, was calculated from the electronic energy of the complex and electronic energies of its subsystems using the BLYP-D3(BJ)/def2-TZVP//BLYP-D3(BJ)/def2-SVP scheme. For
TwinX ⊃ C
60
and
TwinX ⊃ 2C
60
complexes, the interaction energy can be calculated as follows:
The deformation energy, Δ
E
def
, is the amount of energy required to deform the individual fragments from their equilibrium structure to the geometry that they adopt in the complex:
A quantitative energy decomposition analysis [
48
,
49
] was used to investigate the interaction energy in the gas phase within the framework of the Kohn–Sham MO model. Δ
E
int
was decomposed into electrostatic interactions, Pauli repulsion, and attractive orbital interactions, with the addition of the term ∆
E
disp
to account for dispersion correction:
The term ∆ V elstat usually refers to the classical electrostatic interactions between the unaltered charge distributions of the prepared fragments. The Pauli repulsion, ∆ E Pauli , is responsible for any steric repulsion and consists of the destabilizing interactions between occupied orbitals. The orbital interactions, ∆ E oi , account for electron-pair bonding, charge transfer interactions, and polarization. The term ∆ E disp accounts for the dispersion corrections [ 50 ].
Exciton delocalization and charge transfer in donor–acceptor complexes were quantitatively analyzed using the transition density [
51
,
52
–
53
]. The analysis was carried out in the more convenient Löwdin orthogonalized basis. The matrix
λ
C
of orthogonalized MO coefficients is obtained from the coefficients
C
in the original basis
λ
C
=
S
1/2
C
, where
S
is the atomic orbital overlap matrix. The transition density matrix
T
0
i
for an excited state
Φ
i
constructed as a superposition of singly excited configurations (where an occupied MO
ψ
i
is replaced by a virtual MO
ψ
a
) is computed,
A key quantity
The weights of local excitations on donor (
D
) and acceptor (
A
) are Ω(
D
,
D
) and Ω(
A
,
A
). The weight of electron transfer configurations
D
→
A
and
A
→
D
is represented by Ω(
D
,
A
) and Ω(
A
,
D
), respectively. The index Δ
q
, which describes charge separation and charge transfer between
D
and
A
, is calculated as
A COSMO-like polarizable continuum model [
54
,
55
–
56
] in the monopole approximation was used to estimate the equilibrium solvation energy
where
f
(ε) is the dielectric scaling factor,
The charge on atom X in the excited state Φ
i
,
The non-equilibrium solvation energy for excited state ψ
i
can be calculated as [
58
]:
In Eq. ( 10 ), n 2 is the refraction index squared, ε is the optical dielectric constant of the medium, and the vector Δ describes the change of atomic charges in the molecule by excitation in terms of atomic charges; see Eq. ( 9 ).
The nonadiabatic electron transfer rate,
k
ET
, can be calculated using the electronic coupling squared,
V
2
, and the Franck–Condon weighted density of states (FCWD):
The Marcus expression is derived for the high-temperature condition,
An effective value of the Huang–Rhys factor S is estimated from the internal reorganization energy λ i as
The fragment charge difference method (FCD) [
60
,
61
] was used to derive the coupling between diabatic states of interest from related adiabatic excited states computed using TDA approach [
64
]. Within the two-state model, the D–A coupling reads:
The reorganization energy was computed at the BLYP-D3(BJ)/def2-SVP level. It is usually divided into two parts,
λ
=
λ
i
+
λ
s
, including the internal and solvent terms. The solvent reorganization energy is the amount of energy required to move solvent molecules from their initial-state location to their CT state location without charge transfer. The
λ
s
for a particular CT states was computed as a difference between the equilibrium (
E
eq
, see Eq.
8
) and non-equilibrium (
E
neq
, see Eq.
10
) solvation energies for states of interest. The internal reorganization energy
λ
i
is the energy of structural changes that occur when donor/acceptor fragments go from initial-state geometries to final-state geometries.
A unique peanut-like topology of the studied molecules as well as their ability to form inclusion complexes with fullerenes both in solution and in the solid state encouraged us to study in detail the binary and ternary complexes of the
TwinX
with one or two
C
60
fullerene molecules. To estimate the stability of the complexes, the interaction energy (∆
E
int
) between the twin nanorings and fullerene was calculated. The obtained values for mono (
Twin1 ⊃ C
60
,
Twin2 ⊃ C
60
, and
Twin3 ⊃ C
60
) and bis (
Twin1 ⊃ 2C
60
,
Twin2 ⊃ 2C
60
, and
Twin3 ⊃ 2C
60
) adducts are shown in Table
1
. As previously demonstrated for similar systems, dispersion interactions play a significant role in the stability of such complexes [
65
,
66
]. To confirm this, we used a Morokuma-type method of energy decomposition analysis (EDA) [
67
,
68
–
69
].
EDA results for binary (
Twin1 ⊃ C
60
,
Twin2 ⊃ C
60
and
Twin3 ⊃ C
60
) and ternary (
Twin1 ⊃ 2C
60
,
Twin2 ⊃ 2C
60
and
Twin3 ⊃ 2C
60
) complexes Complex Interaction scheme[a] Energy terms[b], kcal/mol ∆ ∆ ∆ ∆ ∆ Mono adduct Type 1 86.5 − 35.0 (25%) − 18.0 (13%) − 87.9 (62%) − 54.4 Type 1 77.4 − 32.4 (26%) − 16.5 (13%) − 76.7 (61%) − 48.2 Type 1 71.6 − 30.4 (26%) − 15.3 (13%) − 70.4 (61%) − 44.6 Bis adduct Type 1 96.4 − 40.1 (27%) − 20.1 (13%) − 90.5 (60%) − 54.3 Type 2 96.8 − 39.4 (26%) − 20.2 (13%) − 91.1 (61%) − 53.9 Type 3 0.03 − 0.02 − 0.01 − 0.53 − 0.53 − 108.2 Type 1 77.3 − 32.0 (26%) − 16.5 (13%) − 76.7 (61%) − 47.9 Type 2 80.3 − 32.9 (26%) − 16.9 (13%) − 77.2 (61%) − 46.7 Type 3 0.00 − 0.01 0.00 0.00 − 0.01 − 94.6 Type 1 71.5 − 29.6 (26%) − 15.4 (13%) − 70.6 (61%) − 44.1 Type 2 71.2 − 30.7 (26%) − 15.3 (13%) − 70.5 (61%) − 45.3 Type 3 0.00 − 0.01 0.00 0.00 − 0.01 − 89.4Table 1
As can be seen in Table 1 , the destabilizing term (Pauli repulsion) decreases when moving from Twin1 ⊃ 2C 60 to Twin3 ⊃ 2C 60 complex. The dispersion term dominates the binding forces (electrostatic, orbital, and dispersion interactions) and accounts for approximately 61% of the total. It is followed by the electrostatic (about 26%) and orbital (about 13%) interactions. Similar behavior has been observed in other van der Waals complexes of C 60 with cycloparaphenylenes [ 66 ].
We compared the geometries of the complexes to explain the observed differences. The guest molecule ( C 60 ) is the same in all complexes. Its effective radius R eff (the mean distance from center to each atom) is 5.150 Å. The size of the host molecules, however, differs significantly. Let us first compare the mono adduct complexes Twin1 ⊃ C 60 , Twin2 ⊃ C 60 , and Twin3 ⊃ C 60 . As seen in Fig. 1 , both wings of Twin1 are in the shape of a circle, whereas Twin2 and Twin3 have a distinct ellipsoid shape with wings extending away from the linker. The structures of the double nanohoops and their geometrical parameters are provided in Fig. S1, SI. In Twin1 ⊃ C 60 , the C 60 fullerene is surrounded by ten phenylene units at a distance of 6.879 Å. The comparison of Twin1 ⊃ C 60 with the [10]CPP ⊃ C 60 complex, in which both units are perfectly size matched, demonstrates a high similarity of the effective radius ( R eff = 6.867 Å in [10]CPP ⊃ C 60 ), and the interaction energy (Δ E int = − 56.8 kcal/mol for [10]CPP ⊃ C 60 ). In both Twin2 ⊃ C 60 and Twin3 ⊃ C 60 , C 60 interacts with only eight phenylene units at the effective distance of about 6.9 Å (Fig. S2, SI). Thus, the weaker interactions in Twin2 ⊃ C 60 and Twin3 ⊃ C 60 can be explained by the smaller number of phenylene units available for interaction with C 60 . The higher Δ E int values in Twin1 compared to Twin3 are consistent with the larger association constants of the former.
The relationship between nanohoop size and the interaction energy found for the mono adduct is also valid for Twin1 ⊃ 2C 60 , Twin2 ⊃ 2C 60 , and Twin3 ⊃ 2C 60 . The formation energy of the ternary complexes can be expected to be superadditive due to the additional interactions between two fullerenes, similar to that observed in the complexes of fullerenes with other curved carbon nanostructures [ 70 , 71 ]. However, the distance between fullerenes in these complexes is too large (13.590 Å, 27.047 Å, and 26.694 Å for Twin1 ⊃ 2C 60 , Twin2 ⊃ 2C 60 , and Twin3 ⊃ 2C 60 ) for such interactions (Fig. S3, SI). This is confirmed by the calculated ∆ E int (Table 1 ), which is less than − 0.5 kcal/mol.
The non-covalent interaction index (NCI) was used to describe the topology of the host–guest interactions [ 72 ]. The shape of NCI isosurfaces clearly demonstrates the difference between the intermolecular interactions in Twin1 ⊃ C 60 , Twin2 ⊃ C 60 , and Twin3 ⊃ C 60 . In the case of Twin1 ⊃ C 60 , the isosurface is distributed fairly uniformly between C 60 and one of the nanohoops. In turn, in Twin2 ⊃ C 60 and Twin3 ⊃ C 60 , the isosurface surrounds the fullerene only partially, indicating interactions with eight out of ten phenyl rings. Figures S4–S7 in SI provide the NCI isosurfaces and reduced density gradient (RDG) plots for mono and bis adducts.
The topological study based on Bader's Atoms in Molecules theory [ 43 ] provides more details regarding the host–guest interactions in the complexes. Table S1 in SI show the electron density, its Laplacian, and other topological parameters at bond critical points (BCPs). The analysis confirmed that the interactions between the fragments in both binary and ternary complexes correspond mostly to π···π interactions. When moving from Twin1 to Twin3 -based complexes, the number of BCPs notably decreases. For example, the number of BCPs between the nanohoops and C 60 in Twin1 ⊃ C 60 , Twin2 ⊃ C 60 , and Twin3 ⊃ C 60 complexes is 22, 16, and 11, respectively. Only a few of them correspond to CH···π interactions. The number of BCPs and characteristics of the electron density at these points are in agreement with the interaction energy values of the complexes. The nature of non-covalent interactions in ternary Twin1 ⊃ 2C 60 , Twin2 ⊃ 2C 60 , and Twin3 ⊃ 2C 60 complexes is similar to the nature in binary complexes. Figures S8 and S9 in SI show the QTAIM molecular graphs for each complex studied.
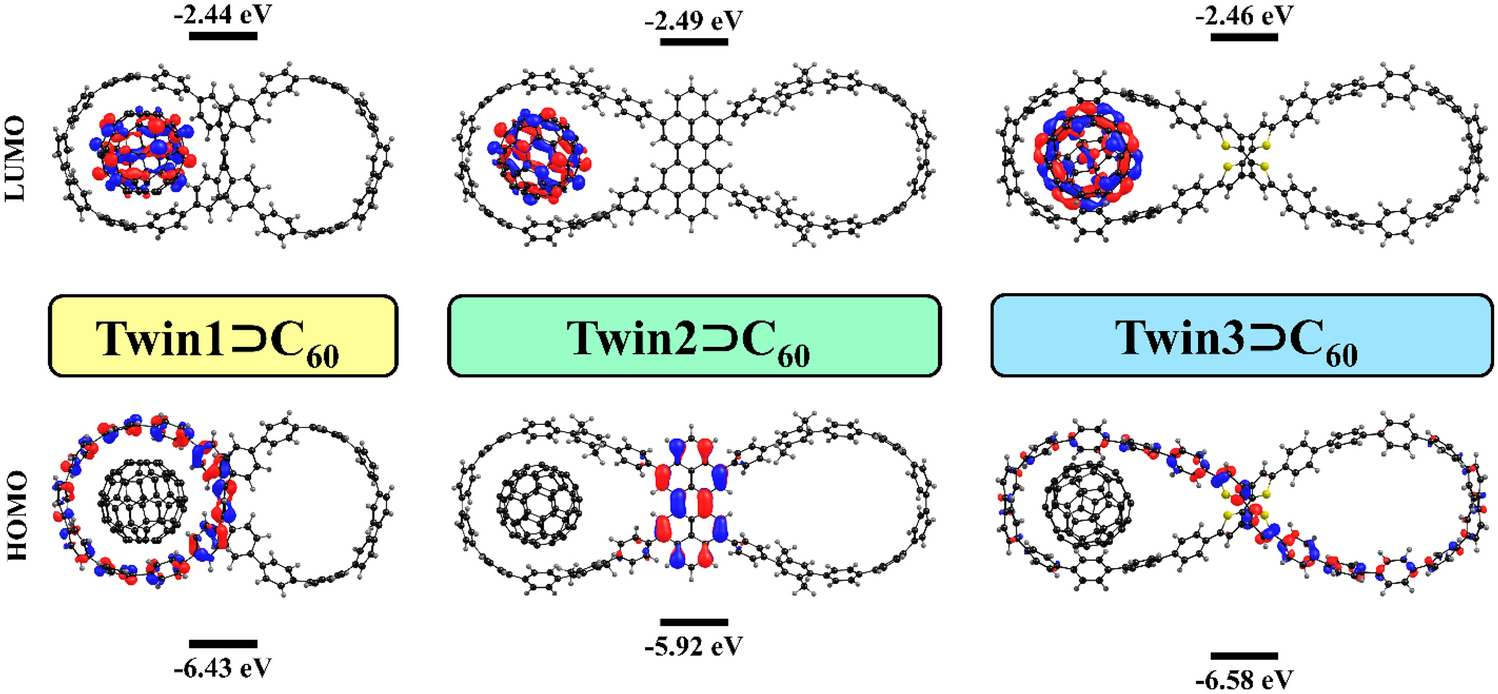
Let us compare the orbital energies of the host and guest molecules and their complexes. All complexes have similar HOMO and LUMO energies, as shown in Fig.
2
. The HOMO is localized on the nanohoop, while the LUMO is localized on the fullerene unit (Fig.
3
). The HOMO energies of nanohoops remain almost unchanged (within 0.1 eV) upon complex formation. A shift in the LUMO energy of the fullerene is less than 0.2 eV. These small differences in orbital energies when going from the individual molecules to their complex indicate the absence of notable changes in the fragment structures as well as the lack of charge separation between the units in the ground state (GS), in accordance with the low contribution of the Δ
E
oi
term to the bonding interaction (vide supra).
Structure and HOMO/LUMO energy levels of the studied binary and ternary complexes Localization of Kohn–Sham HOMO and LUMO in the
TwinX ⊃ C
60
complexesFig. 2
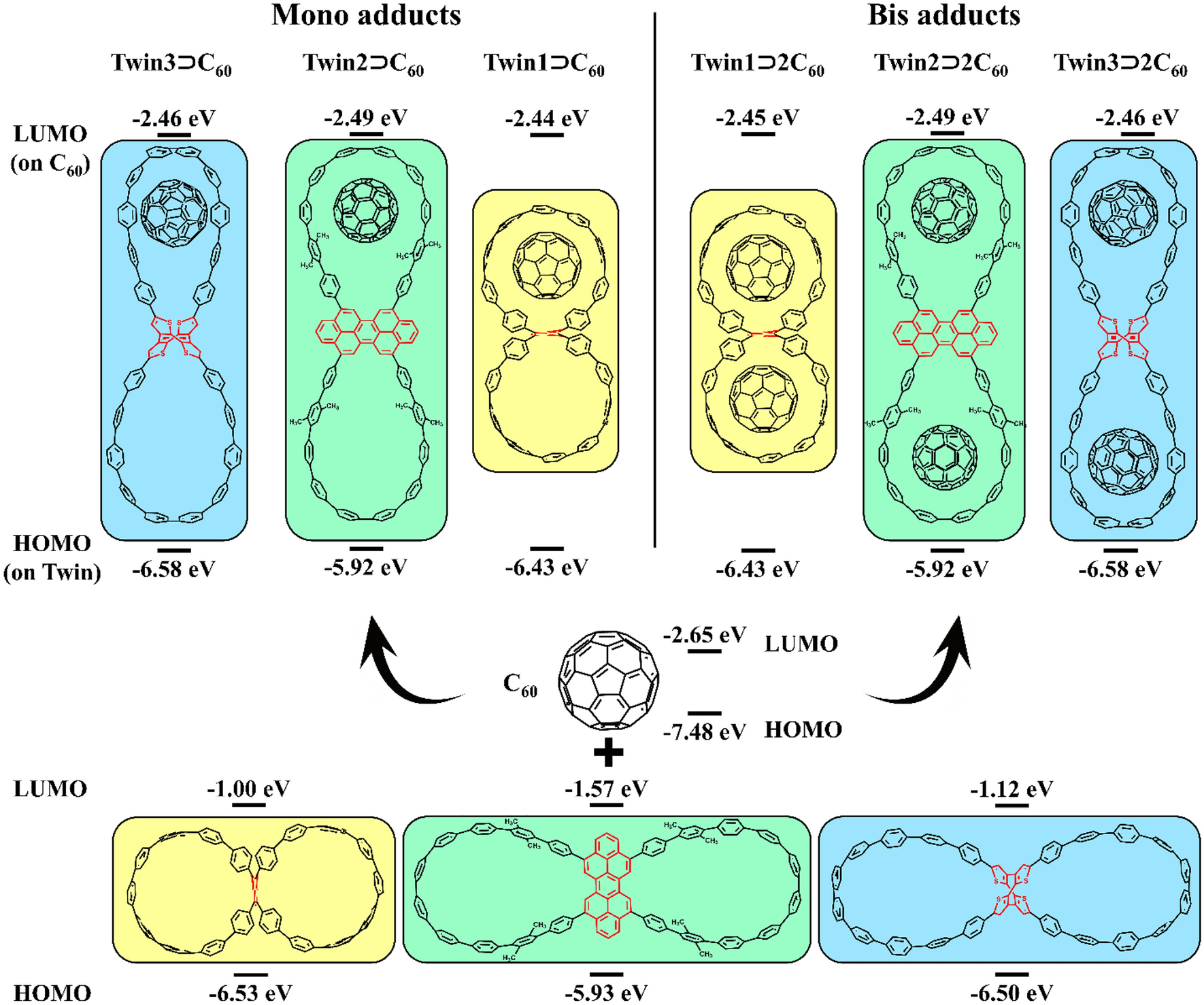
Fig. 3
The deformation energy (Δ E def ) was calculated to quantify structural changes. The greatest Δ E def = 6.5 kcal/mol was found for Twin1 ⊃ C 60 , with a dominant contribution from the nanohoop. In other complexes, both fullerene and nanohoop fragments make similar contribution (Table S2, SI). As seen in Fig. S10, the deformations in Twin1 ⊃ C 60 are mainly associated with changes in the twist angle of the phenylene units of the nanohoops. In Twin3 ⊃ C 60 , the changes in the dimensions of the nanohoop wings are caused by the interaction with fullerene. Twin2 ⊃ C 60 has the smallest Δ E def , which correlates with the smallest root-mean-square deviation (RMSD) of atomic positions. Population analysis performed with several charge schemes shows no significant charge transfer between the host and guest molecules in the complexes (Table S3, SI).
PET properties of the nanohoop are dependent on both their structural (size of wing and type of linker) and electronic (HOMO energy) characteristics. Each system was divided into two (for
TwinX ⊃ C
60
) or three (for
TwinX ⊃ 2C
60
) fragments: nanohoop, acting as an electron donor; and
C
60
fullerene, acting as an electron acceptor. The lowest 80 excited states were investigated for their contribution to charge transfer and exciton delocalization (Table
2
).
Excitation energies (
E
x
, eV), main singly excited configuration (HOMO(H)–LUMO(L)) and its weight (W), oscillator strength (f), extent of charge transfer (CT, e) or localization of exciton (X), and difference in dipole moment with respect to the ground state (∆μ, D) between GS and CT state computed for binary (
TwinX ⊃ C
60
) and ternary (
TwinX ⊃ 2C
60
) complexes on nanohoops in the gas phase LEGuest ( Ex 2.495 2.501 2.494 2.479 2.501 2.493 Transition (W) H-5 – L + 1 (0.54) H-5 – L + 2 (0.39) H-7 – L + 1 (0.28) H-5 – L + 1 (0.14) H-7 – L + 2 (0.38) H-7 – L + 5 (0.33) f < 0.001 < 0.001 < 0.001 < 0.001 < 0.001 < 0.001 X 0.933 0.943 0.935 0.926 0.946 0.931 LEHost (Nanohoop Ex 3.220 2.857 3.416 3.240 2.863 3.424 Transition (W) H – L + 3 (0.47) H – L + 3 (0.88) H-1 – L + 8 (0.11) H – L + 6 (0.32) H – L + 6 (0.86) H-2 – L + 14 (0.13) f 0.008 1.179 0.037 0.003 1.056 0.020 X 0.909 0.968 0.810 0.874 0.949 0.872 Most absorptive (MA) transition Ex 3.698[a] 3.778 3.498 3.641 3.801 3.586[a] Transition (W) H – L + 9 (0.29) H – L + 12 (0.15) H-1 – L + 6 (0.26) H – L + 12 (0.08) H – L + 13 (0.14) H – L + 14 (0.08) f 2.952 4.116 5.472 3.006 4.091 6.278 Localization X 0.770 0.978 0.975 0.849 0.939 0.441 CT1 ( Ex 2.615 2.800 2.757 2.686 2.800 2.752 Transition (W) H – L (0.85) H-2 – L (0.72) H-2 – L + 1 (0.25) H – L + 4 (0.45) H-1 – L (0.71) H-1 – L + 1 (0.31) f 0.002 0.001 0.018 0.002 0.001 0.012 CT 0.944 0.950 0.956 0.973 0.953 0.959 ∆μ 4.45 13.67 12.29 1.71 14.27 11.70 CT2 ( Ex n/f [b] 2.688 3.523 n/f [b] 2.688 3.381 Transition (W) H – L (0.81) H-1 – L + 2 (0.25) H – L (0.81) H-3 – L + 3 (0.23) f 0.003 0.019 0.003 0.009 CT 0.984 0.941 0.985 0.978 ∆μ 56.65 39.91 56.71 26.91Table 2
Three types of the excited states were identified: (1) locally excited (LE) states are those in which excitation is localized on either the fullerene (LE Guest ) or the nanohoop (LE Host ) and charge transfer between the units is less than 0.1 eV (CT < 0.1 e ); (2) charge transfer (CT) states with high charge separation (CT > 0.8 e ); and (3) mixed states, which combine the contributions of both LE and CT states (0.1 e < CT < 0.8 e ).
The lowest 80 vertical singlet excitation energies of the binary complexes in the gas phase range from 2.50 to 4.37 eV. In all complexes, the lowest-lying excited states are localized on the fullerene unit (LE
Guest
). Excited states localized on the twin nanohoops (LE
Host
) are higher in energy compared to LE
Guest
. The lowest LE
Host
state was found in
Twin2 ⊃ C
60
, with a small energy difference between LE
Guest
and LE
Host
of 0.36 eV. For
Twin1 ⊃ C
60
and
Twin3 ⊃ C
60
,
such a difference is 0.73 and 0.92 eV correspondingly (Table
2
). Among the excited states studied, two types of CT states have been found. Both are generated by electron transfer from
TwinX
to
C
60
and can be denoted as
TwinX
+
⊃ C
60
−
. For the first CT type, the dipole moment difference between GS and CT states ranges between 4 and 14 D, while the second type of CT is characterized by a significantly stronger dipole moment difference (56.7 and 39.9 D in
Twin2 ⊃ C
60
and
Twin3 ⊃ C
60
, respectively). It is important to note that the CT2 state was not found in
Twin1 ⊃ C
60
. Since
Twin1
has the benzene ring linker, unlike the other complexes with extended π-conjugated linkers, the formation of the CT2 state could be related to electron transfer from the linker to the fullerene. The low-lying CT2 state is not observed in
Twin1 ⊃ C
60
complex, because the HOMO energy of the benzene linker is significantly lower compared to the HOMOs of peropyrene and cyclooctatetrathiophene. Moreover, the higher energy of CT2 state in
Twin3 ⊃ C
60
can be explained by the lower HOMO energy of cyclooctatetrathiophene fragment compared to CPP fragment (see Fig.
2
and Table S4, SI). We repeated the investigation of the excited states dividing the
TwinX ⊃ C
60
complexes into three fragments (fullerene, linker, and CPP wings) to gain further insight. We have found that in the
Twin2 ⊃ C
60
complex, electron transfer in CT2 occurs exclusively from the linker to
C
60
. The two types of CT states in
Twin2 ⊃ C
60
are shown in Fig.
4
.
NTO orbitals for two types of CT states in
Twin2 ⊃ C
60Fig. 4
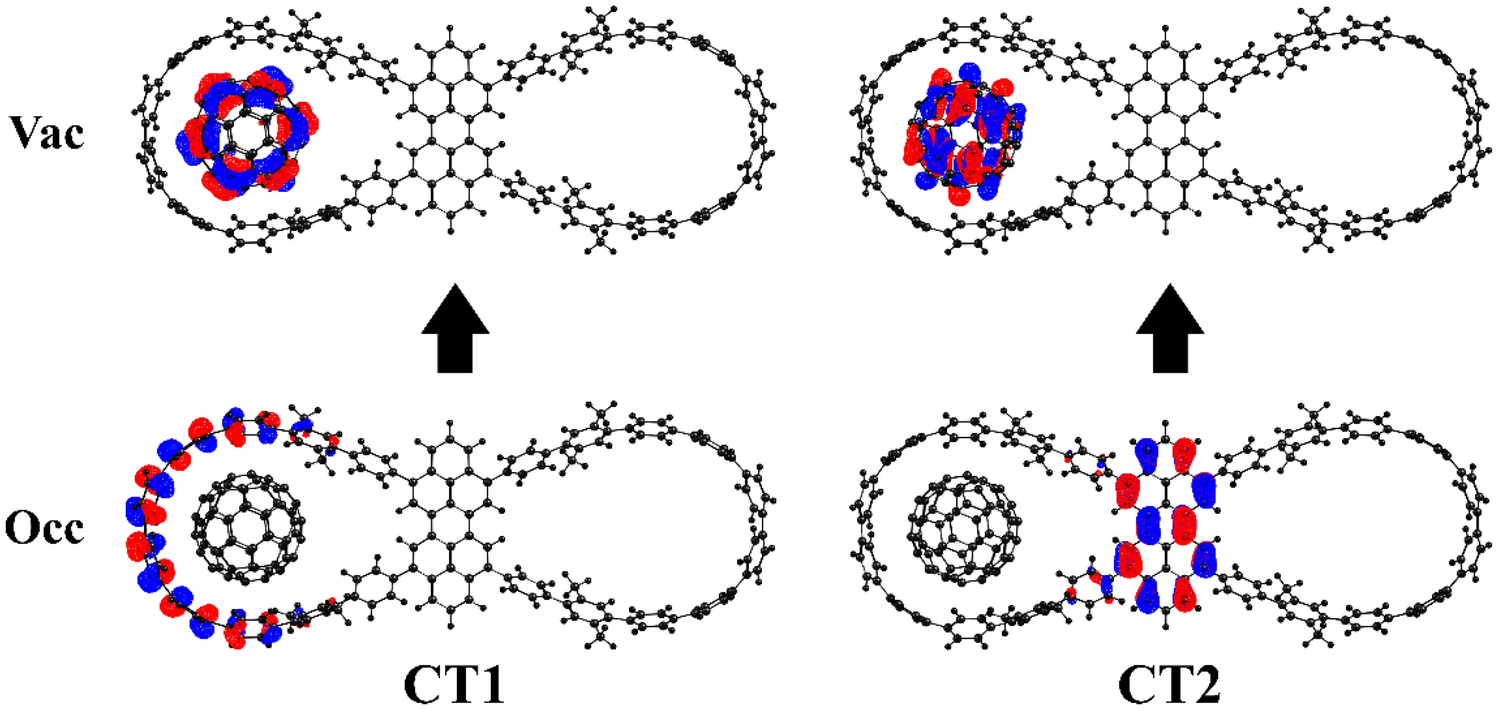
The nature of CT1 and CT2 states in Twin3 ⊃ C 60 is similar to that in Twin2 ⊃ C 60 . Thus, CT1 is generated by electron transfer from the CPP wing of TwinX to the fullerene unit, while the case of CT2 a dominant involvement of the linker in electron transfer is observed. As expected, no CT2 state was found in Twin1 ⊃ C 60 . Figures S11–S13 in SI depict natural transition orbitals (NTOs) for the LE and CT states.
Behavior of the ternary complexes TwinX ⊃ 2C 60 is similar to the binary ones. Due to a higher density of states, the energies of the lowest 80 excited singlet states in the systems range from 2.48 to 3.80 eV. The energies of both LE Guest and LE Host states in the ternary and binary complexes are very similar. A qualitative difference between Twin1 ⊃ C 60 and Twin1 ⊃ 2C 60 was found for the LE Host states (see Figs. S11 and S14, SI). In Twin1 ⊃ C 60 , LE Host is only on one CPP wing, while in Twin1 ⊃ 2C 60 the LE Host is delocalized over the entire Twin1 molecule. In the binary system, two CPP wings are not structurally similar—one contains fullerene and the other does not. Interaction with C 60 increases the energy of the orbital located at this CPP, resulting in LE Host being located only on this wing. In Twin1 ⊃ 2C 60 , both CPP wings are nearly equivalent and the state is delocalized over them.
In Twin2 ⊃ 2C 60 and Twin3 ⊃ 2C 60 , two types of CT states can also be distinguished. In Twin2 ⊃ 2C 60 and Twin 2 ⊃ C 60 their characteristics are very similar, whereas in Twin3 ⊃ 2C 60 the energy of the CT2 state is 0.2 eV lower compared to Twin3 ⊃ C 60 . Analysis of the CT2 states in Twin3 ⊃ C 60 and Twin3 ⊃ 2C 60 complexes revealed that its lower energy in Twin3 ⊃ 2C 60 is associated with greater delocalization of the occupied NTO along the phenyl rings of CPP (compare Figs. S13 and S16, SI). The NTOs describing LE and CT states in TwinX ⊃ 2C 60 complexes are given in Figs. S14–S16 in SI.
It is widely accepted that solvation has a great influence on both the ground and excited states. While CT states can be highly stabilized or destabilized by the solvent, the influence of solvation on LE states is often minimal. The equilibrium COSMO-like solvation model [ 66 , 73 , 74 ] with dichloromethane (DCM) as the solvent was used to evaluate the effect of the solvent on the excited states. All of the investigated complexes in GS have rather small dipole moments. For binary systems, the dipole moments vary from 0.25 to 0.87 D, whereas for more symmetrical ternary complexes, these values decrease to 0.06–0.13 D. For Twin1 ⊃ C 60 , Twin2 ⊃ C 60 , and Twin3 ⊃ C 60 , the GS solvation energies are −0.77, −0.92, and −1.00 eV, correspondingly. The similarity in the dipole moments results in similar solvation energies. The solvation energies of the ternary analogs differ by no more than 0.05 eV (Table S5, SI).
For the LE
Guest
and LE
Host
states, the overall picture is very similar to the GS state. A comparison of excitation energies computed in the gas phase and in DCM solution shows that the LE transition energies remain almost unchanged, which in turn correlates perfectly with negligibly small changes in the dipole moment when going from GS to LE states. A difference (∆μ) of the dipole moment vector in the LE and GS states in the complexes does not exceed 1 D. The changes in the dipole moment associated with CT states are significantly larger compared to the LE states. Despite the fact that both types of CT have a similar electronic structure, their response to solvation can be different. Indeed, the solvation energies calculated for the CT states are in good agreement with the corresponding ∆μ values. For CT1 states characterized by a moderate ∆μ, the difference in solvation energies of the GS and CT states ranges from 0.18 to 0.30 eV. The CT2 states in
Twin2
and
Twin3
based complexes show significantly larger ∆μ values and, as a result, larger solvation energies. Figure
5
displays the energies of the GS, LE, and CT states in the gas phase and DCM. Detailed data for all complexes is collected in Table S5 in SI.
Energies of the LE and CT states (in eV) computed for the binary (
TwinX ⊃ C
60
) and ternary (
TwinX ⊃ 2C
60
) complexes in vacuum (VAC) and dichloromethane (DCM). In the figure, LE1 stands for LE
GuestFig. 5
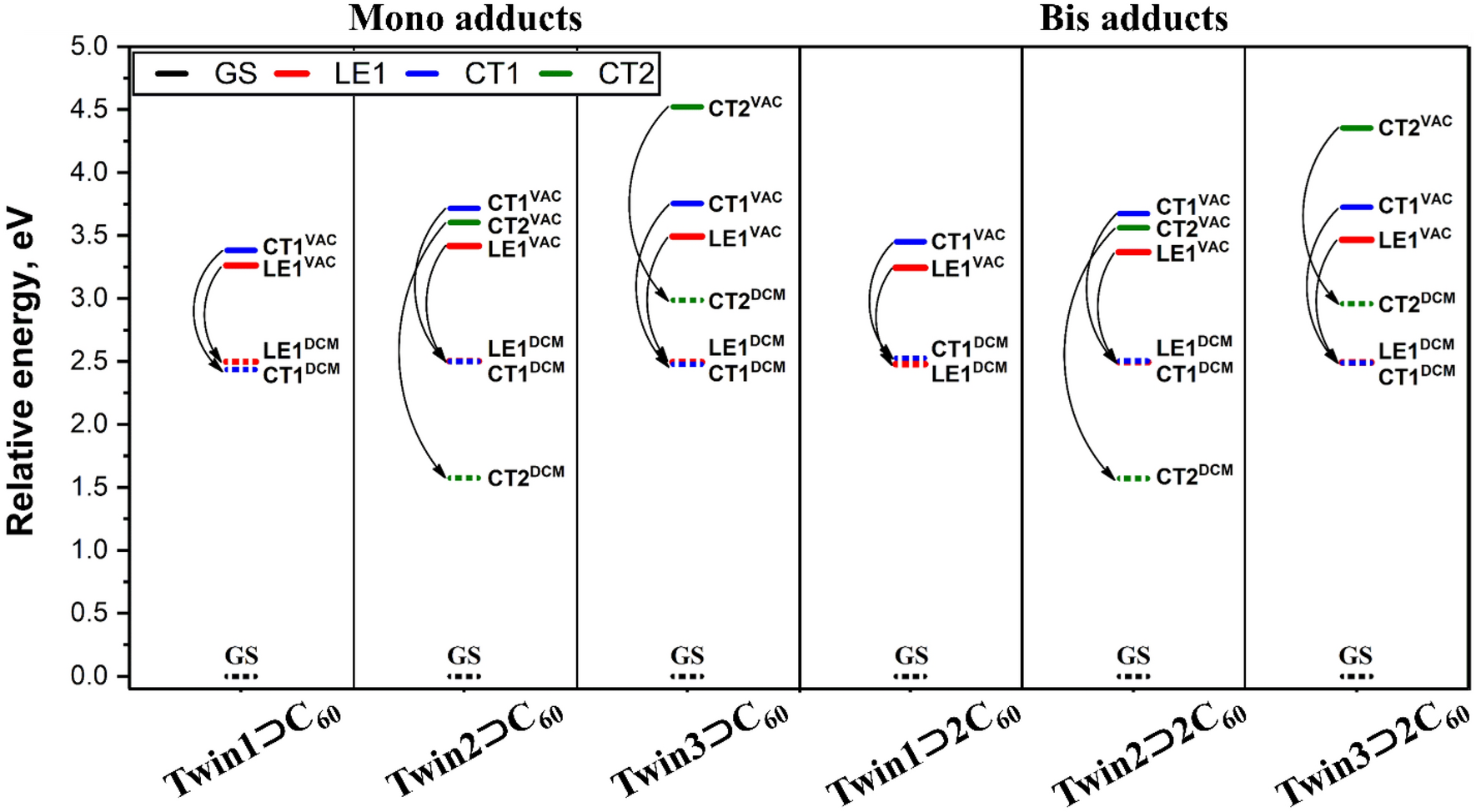
In all cases, the solvent stabilization of the CT1 state is enough to balance the energies of the LE Guest and CT1 states. The solvation of the Twin2 -based complexes was found to lower the energy of the CT2 state making it by almost 1 eV smaller than that of the LE Guest . In the case of Twin3 -based complexes, the higher energy of the CT2 state in vacuum and its smaller solvation energy do not allow this state to become sufficiently low. Simulated absorption spectra for the complexes are given in Fig. S17 in SI.
Because all CT states in the complexes have very weak oscillator strengths, they cannot be populated directly by light absorption. On the other hand, locally excited states populated by strongly absorbing transitions dissipates to the lowest-lying LE state by a non-radiative decay channel. In turn, the last state can decay to a lower CT states by electron transfer between the donor and acceptor sites.
Semi-classical approach proposed by Ulstrup and Jortner [
62
,
63
] was used to estimate the rates of charge separation (
k
CS
) and charge recombination (
k
CR
) processes. According to this method, an effective vibrational mode is used to describe the intramolecular relaxation associated with ET. In addition, the rate is controlled by the electronic coupling between the initial and final states,
V
ij
, solvation reorganization energy,
λ
s
, Gibbs free energy, ∆
G
0
, and effective Huang-Rhys factor,
S
eff
. We have previously shown that changing the effective frequency from 1400 to 1800 cm
−1
does not lead to a significant change in the charge separation rate [
75
,
76
]. Thus, the effective frequency of 1600 cm
−1
, which corresponds to the stretching of C=C bonds, was used to estimate the rates. Table
3
includes the computed parameters for charge separation processes in DCM solvent for the complexes of interest.
Charge separation rates (
k
CS
, s
−1
), Gibbs energy (∆
G
0
, eV), electronic coupling (|
V
ij
|, eV), solvent (
λ
s
) and internal (
λ
i
) reorganization energy (eV), Huang–Rhys factor (
S
eff
) and activation energy barrier (∆
E
a
, eV) for mono (
TwinX ⊃ C
60
) and bis (
TwinX ⊃ 2C
60
) adducts computed in DCM Complex ∆ | Reorg. energy ∆ τ Mono adducts CT1 −0.063 1.33·10–3 0.129 0.192 0.650 0.022 2.93·1010 0.03 CT1 CT2 −0.001 −0.927 4.62·10–3 9.92·10–4 0.132 0.200 0.253 0.724 0.665 1.008 0.063 0.009 6.21·1010 2.06·1010 0.02 0.05 CT1 CT2 −0.017 0.492 3.46·10–3 3.36·10–3 0.145 0.183 0.226 0.419 0.726 0.923 0.049 0.495 6.46·1010 [1.23·103] 0.02 − Bis adducts CT1 0.048 1.08·10–3 0.131 0.162 0.660 0.068 3.45·109 0.29 CT1 CT2 0.009 −0.924 4.05·10–3 7.36·10–4 0.141 0.211 0.252 0.726 0.711 1.064 0.068 0.009 3.96·1010 1.16·1010 0.03 0.09 CT1 CT2 −0.003 0.466 2.68·10–3 2.75·10–3 0.143 0.194 0.213 0.344 0.721 0.978 0.052 0.476 3.49·1010 [1.89·103] 0.03 −Table 3
As can be seen in Table 3 , the reorganization energy for the ET reactions varies significantly. The LE Guest → CT1 charge separation process in all complexes is characterized by moderate internal reorganization energies (from 0.13 to 0.15 eV) and solvation reorganization energies (from 0.16 to 0.25 eV). This process occurs in the normal Marcus regime (|∆ G 0 |< λ) [ 59 , 77 ] on the sub-nanosecond timescale. In contrast, ET parameters that control generation of the CT2 states are quite different. The LE Guest → CT2 charge separation process shows significantly higher reorganization energies. In the Twin2 - and Twin3 -based complexes, the solvent reorganization energy for this reaction is more than twice as high as for LE Guest → CT1 process. In Twin2 ⊃ C 60 and Twin2 ⊃ 2C 60 complexes, LE Guest → CT2 reaction is nearly barrierless. The characteristic time (τ) was found to be 0.05 and 0.09 ns, respectively. The CT2 charge separation in Twin3 ⊃ C 60 and Twin3 ⊃ 2C 60 is characterized by a strong positive Gibbs energy and thus this reaction is unlikely to occur.
Typically, excited CT states decay to the ground state by charge recombination. The effect of internal geometry reorganization on ∆G
0
is rather small for large π-conjugated systems, such as the complexes studied, and can be safely ignored [
65
,
78
]. The computed charge recombination rates (
k
CR
) are provided in Table
4
. Charge recombination rates of CT2 for
Twin3 ⊃ C
60
and
Twin3 ⊃ 2C
60
complexes were not considered because of their low probability.
Charge recombination rates (
k
CR
, s
−1
), Gibbs energy (∆
G
0
, eV), electronic coupling (|
V
ij
|, eV), solvent (
λ
s
) and internal (
λ
i
) reorganization energy (eV), and Huang–Rhys factor (
S
eff
) for mono (
TwinX ⊃ C
60
) and bis (
TwinX ⊃ 2C
60
) adducts computed in DCM ∆ | Reorg. energy Mono adducts CT1 − 2.436 7.95·10–3 0.149 0.192 0.751 1.63·103 CT1 CT2 −2.501 −1.575 3.43·10–2 1.35·10–2 0.140 0.189 0.253 0.724 0.706 0.953 2.10·104 1.80·1011 CT1 −2.478 2.69·10–2 0.151 0.226 0.761 2.25·104 Bis adducts CT1 −2.527 5.46·10–3 0.155 0.162 0.781 1.90·102 CT1 CT2 −2.504 −1.571 4.65·10–2 1.29·10–2 0.148 0.199 0.252 0.726 0.746 1.003 6.24·104 1.90·1011 CT1 −2.490 1.29·10–2 0.155 0.213 0.781 4.52·103Table 4
In contrast to charge separation, the charge recombination reactions take place in the deep inverted Marcus region (|∆G 0 |≫ λ) [ 59 , 77 ]. The decay of CT1 states is significantly slower than their generation. However, the charge recombination of CT2 states in Twin2 ⊃ C 60 and Twin2 ⊃ 2C 60 complexes is an order of magnitude faster than the charge separation reaction. The fast decay of CT2 states prevents their experimental observation. Note that the charge recombination rates assessments are rather qualitative and should be treated with caution.
Using the TD-DFT approach, we investigated the ground and excited-state properties of the host–guest complexes based on three experimentally obtained double nanohoops and C 60 fullerene. The energy decomposition analysis revealed that the stability of the complexes is determined by dispersion interactions between the host and guest molecules. Two types of CT states were found: CT1, generated by electron transfer from CPP to C 60 , and CT2, in which an electron is transferred from the linker to C 60 . Efficient population of the CT2 state is possible only for Twin2 -based complexes due to the peropyrene linker. However, the rapid deactivation of this state is a significant disadvantage that prevents its experimental observation. We demonstrated that the efficient photoinduced electron transfer from the CPP fragment to C 60 occurs on sub-nanosecond timescale. Sufficiently slow charge recombination processes found for the CT1 states lead to an efficient separation of electrons and holes, making the complexes attractive candidates for use in organic photovoltaics.
We acknowledge financial support from the Spanish MINECO (Network RED2018-102815-T, project PID2020-113711GB-I00, and Juan de la Cierva contract IJC2019-039846-I to A.J.S.), the Catalan DIUE (2017SGR39), and the University of Girona (POSTDOC-UdG 2021/31 to O.A.S.). This research was supported in part by PLGrid Infrastructure.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.