Graphical Abstract

Hydrogen adsorption was investigated in Li- and K-doped single-walled silicon carbide nanotubes (SWSiCNT) by applying the grand canonical Monte Carlo simulation. Our results depict that hydrogen storage increases as a function of pressure at fixed temperature. We considered seven doping sites on the nanotube. The results indicate that except for some doping sites, hydrogen adsorption in Li- and K-doped SWSiCNT is more than in SWSiCNT without doping. Another observation is that the hydrogen physisorption in K-doped SWSiCNT is more than that in Li-doped SWSiCNT. The hydrogen adsorption in Li- and K-doped single-walled silicon carbide nanotube arrays (SWSiCNTAs) have also been calculated and illustrate that K-doped SWSiCNTAs have more hydrogen adsorptivity than Li-doped SWSiCNTAs which is in contrast to the obtained results in carbon nanotubes arrays. In this study, we plotted the energy of hydrogen adsorption to confirm the adsorption isotherms and then we fit the simulation data into the Langmuir and Langmuir–Freundlich equation. It was found that multi-layer absorptivity occurs prominently more spatially in K-doped SWSiCNT. However in some doped sites, for both Li- and K-doped SWSiCNT, the dominant mechanism was monolayer adsorptivity which was due to low hydrogen adsorption.
The hydrogen potential in adsorption and storage of nanochemical structures has been known for approximately two decades. Interest in the use of hydrogen as one of the most promising energy carriers has led to both political and scientific efforts to increase its applications for industrial, economic and environmental goals [ 1 ].
Since the discovery of carbon nanotubes (CNTs) [ 2 ], extensive studies have been carried out on experimental and computing modeling approaches to investigate the physisorption and storage of small molecules in nanotubes [ 3 – 10 ]. The storage of hydrogen inside carbon nanotubes (CNTs) [ 11 – 13 ], boron nitride nanotubes (BNNTs) and their arrays [ 14 – 17 ] and silicon carbide nanotubes (SiCNTs) [ 18 , 19 ], most of which use molecular simulation procedures, has been of special interest. It should be noted that the US Department of Energy (DOE) has established that the maximum gravimetric capacity of hydrogen adsorption in a hydrogen storage media will be achieved at 9 wt% for the year of 2015 [ 20 ]. So far, many researchers have focused their theoretical and experimental studies on finding more feasible hydrogen adsorbing nanomaterials. In 1999, Chen et al. [ 21 ] showed that Li-doped carbon nanotubes could absorb 20 wt% of hydrogen at 1 atm and 200–400 °C, while K-doped carbon nanotubes could absorb 14 wt% of hydrogen at 1 atm and room temperature. Furthermore, in 2000, Yang revisited the work of Chen et al. [ 22 ] and reported that any moisture contamination drastically decreased the hydrogen storage in alkali-doped carbon nanotubes. In fact, moisture from hydrogen reacts with the alkali atoms on carbon. Consequently, dry hydrogen appears more suitable for adsorption in alkali metal-doped carbon nanotubes.
In 2004, Zhu et al. [ 23 ] investigated hydrogen adsorption in Li- and K-doped graphite using the density functional theory (DFT) at 0 K, which is a thermodynamically favorable adsorption. This clearly demonstrates the optimized geometries of hydrogen adsorption on Li- and K-doped graphite to indicate the charge transfer between the metal atoms and the carbon of graphite, revealing that Li is stronger than K in adsorption on graphite [ 23 ].
In 2005, Hu et al. [ 24 ] studied the effect of Li + - and K + -doping SWCNTs on hydrogen adsorption using a Monte Carlo (MC) simulation. It should be noted that Li + and K + ions were inserted into SWCNTs and the hydrogen adsorption of Li-doping SWCNT and K-doping SWCNT resulted in 4.21 and 3.95 wt%, respectively, which are in good agreement with the experimental results obtained at 100 atm and room temperature [ 24 ].
In 2007, Cho et al. [ 25 ] experimented with hydrogen storage on Li-doped single-walled carbon nanotubes (SWCNTs) using DFT. Their work investigated the mechanism of hydrogen adsorption through Li-doping SWCNTs. They found two possible hydrogen physisorption sites in Li-doping SWCNTs: far from the Li while near the nanotube wall (R1) and near the Li while far from the nanotube wall (R2). Their results emphasized that R2 is more hydrogen adsorbable than R1. Cho et al. [ 25 ] calculated hydrogen adsorption energy for each adsorption site (R1 and R2) in Li-doped SWCNTs as a function of the atomic doping ratio of Li atoms. The adsorption energy in R2 is approximately two times greater than that of pristine SWCNTs, while the adsorption in R1 at moderate temperature and pressures is not different from that of pristine SWCNTs.
In 2009, using DFT, Liu et al. [ 25 ] provided the binding mechanisms that govern hydrogen storage in Li-dispersed SWNTs and illustrated that a configuration of eight Li atoms dispersed at the hollow sites above the hexagonal carbon rings can lead to the extremely high hydrogen storage capacity of 13.45 wt%.
Furthermore, Yuan et al. [ 26 ] presented the properties of hydrogen physisorption in K-doped (15, 15) single-walled carbon nanotube arrays (SWCNTAs) by grand canonical Monte Carlo (GCMC) simulation at 293 and 10 MPa as a function of K-doping schemes of K atoms’ doped sites on SWCNTs. The distance between the tubes (DBT) and the height of K atom from the hexagon center in all cases (H) are 1 and 0.16 nm, respectively, and the ratio of K:C is 1:2. They considered three sketch maps of K-doping: (1) K atoms are uniformly located on the outer surface of SWCNTs ( H = 0.16 nm), denoted by (UH); (2) K atoms are specially located at the hexagon centers of SWCNTs ( H = 0), denoted by (S), and (3) K atoms are specially located above the hexagon centers of SWCNTAs ( H = 0.16 nm), denoted by (SH). The adsorption isotherms of hydrogen physisorption clearly show that the hydrogen storage capacity for the S map is more than that of the SH and UH maps (S > SH > UH) [ 26 ]. They found that the height of K-doped sites affects the SWCNT’s Lennard–Jones (LJ) potential wells and hydrogen adsorption. However, in H = 0.16 nm, whether K was outside or inside the tube, the depth of the LJ potential well became equal, so they considered this height for their simulations [ 26 ]. In 2010, Cheng et al. [ 26 ] continued previous studies on hydrogen physisorption in Li-doped SWCNTA by GCMC simulation [ 20 ]. The investigations of hydrogen adsorption in both Refs. [ 20 , 26 ], were done under the same thermodynamic conditions and using the same structure and size of SWCNTAs. Yuan et al. [ 20 ] and Cheng et al. [ 26 ] both concluded that the doping scheme and SWCNTA’s structure and size were important parameters to reach the DOE’s 2015 target for the hydrogen storage capacity of K- and Li-doped SWCNTA.
Based on the investigations on the influence of alkali atom (Li- and K-) doped SWCNTs on hydrogen adsorption and the higher capability of single-walled silicon carbide nanotubes (SWSiCNT) than SWCNTs on hydrogen physisorption [ 18 , 19 ], in the present paper we have studied the effects of Li- and K-doped SWSiCNT to determine whether they are good candidates for practical DOE target material [ 20 – 22 , 25 , 26 ].
In this work, we first provide a brief review of GCMC and the model that is employed in the simulation method. We then present our results for hydrogen adsorption in Li- and K-doped SWSiCNT and compare these results with those obtained for hydrogen physisorption by Li- and K-doped SWCNTs and SWCNTAs in “ Results and discussion ” section. Finally, “ Conclusions ” rounds off the paper.
In this work, following an approach previously described in the literature [ 10 , 17 , 27 ], we used GCMC simulations to investigate hydrogen adsorption in Li- and K-doped SWSiCNT. It should be noted that some of the previous work considered (15, 15) SWSCNT in their calculations [ 20 , 26 ] and, therefore, we considered this nanotube and H = 0.16 nm ( H is the height of Li and K from outside the nanotube wall) to find the best comparison between the hydrogen adsorption of Li- and K-doped (15, 15) SWSiCNT and Li- and K-doped (15, 15) SWCNT. Since some reports used the DFT theory [ 23 , 25 ] and others inserted alkali atoms inside the SWCNTs [ 24 ], it was not deemed reasonable to compare them with our work and they were thus not considered here. In this paper, the length of (15, 15) SWSiCNT is 4 nm, according to SWCNT in Refs. [ 20 , 26 ].
In GCMC simulation, the configurations are sampled from a grand canonical (GC) ensemble [ 10 , 17 , 27 ] and the periodic boundary conditions (PBC) are applied at Li- and K-doped open end (15, 15) SWSiCNT during movements in GCMC simulation until the number of hydrogen molecules in the simulation cell comes to equilibrium [ 10 , 17 , 27 ]. The spherical cutoff distance is set a little smaller than half of the simulation cell [ 10 , 17 , 27 ]. Indeed, for achieving a fixed simulation cell, three types of operations that are generated from a Markov chain are moving, creation and deletion, which are used with equal probability to add a hydrogen molecule inside the simulation cell. The difference between the total potential energy before and after the creation operation must be calculated (Eq. 1 ) and then, according to the assumed acceptance probability (Eq. 2 ), this operation will be accepted or rejected [ 15 , 27 , 28 ]. where r = initial position, r ′ = final random position and P is the probability value [ 28 ].
In GCMC simulation, hydrogen–hydrogen, hydrogen–silicon, hydrogen–carbon, hydrogen–potassium and hydrogen–lithium molecular interactions are considered as spherical Lennard–Jones (LJ) pair potential models which are described by the following Eq. (
3
) [
27
]:
where
ɛ
ij
and
σ
ij
are the energy and the length parameters in the LJ potential and
r
ij
denotes the distance between the centers of
i
and
j
particles. In simulation, the parameters of
ɛ
ij
and
σ
ij
are cross interaction parameters, which are derived from the Lorentz–Berthelot combining rules [
10
,
15
,
17
,
27
,
28
]. Table
1
presents the LJ potential parameters of hydrogen as a fluid; silicon and carbon as SWSiCNT atoms; and Li and K as alkali atoms.
The LJ parameter of hydrogen, silicon, carbon, potassium and lithium that have been considered in this work Atoms C [nanotube] Ref. [ Si [nanotube] Ref. [ K [doped atom] Ref. [ Li [doped atom] Ref. [ H2 Ref. [ LJ parameter 0.340 0.383 0.4115 0.2728 0.2958 43.308 202.429 421.0 567.0 36.7Table 1
In our GCMC simulation, each run consists of 2.5 × 10 7 GCMC moves. The first 1.25 × 10 7 moves are considered as equilibrations which have been discarded, and the last 1.25 × 10 7 moves were used for calculating the ensemble averages of the thermodynamics parameters. The multi-purpose simulation code of the molecular simulation package was used for all simulations [ 28 ]. The Li- and K-doped SWSiCNT have rigid structures, and no geometry variation of the adsorbent (here, adsorbent is doped nanotube) is considered, since the induced geometric variation of doped nanotube can be neglected by hydrogen molecules at room temperature. Finally, the isotherms of hydrogen adsorption are plotted in the doped nanotube at 293 K.
First of all, we considered (15, 15) SWSiCNT and the doping sites on it to plot the hydrogen adsorption isotherms from running GCMC simulations. We found seven sites on this nanotube to dope.
Site 1: the top of all Si and C atoms and the middle of all Si–C bonds.
Site 2: only the top of all Si atoms.
Site 3: only the top of all C atoms.
Site 4: only the top of the middle of all Si–C bonds.
Site 5: only the top–middle of hexagons.
Site 6: the top of all Si and C atoms.
Site 7: only the middle of the hexagons.
It should be noted that in site 5, Li or K as the doping atoms has a height on top of the middle of the hexagons ( H = 0.16 nm), while in site 7, Li or K is directly in the middle of the hexagons without H = 0. To compare the concentration of doping atoms (Li and K atoms), we also calculated the GCMC simulations with consideration to these seven sites, when the full doping position of the sites were occupied by Li or K atoms. We then continued our investigations on the same sites while half of the doping positions were occupied by Li or K atoms. This allowed us to illustrate the concentration of the doping atom on hydrogen adsorption in SWSiCNT. To simplify the expression of these situations, we have shown the full sites filled with Li or K atoms with symbols 1–7 as 1f–7f and the half sites with 1h–7h.
As a result, after running the simulations and obtaining data using GCMC calculations on these 14 situations (seven of them with fully occupied positions and the seven other with half-occupied positions with Li or K atoms), we were able to plot the adsorption isotherms for hydrogen inside Li- and K-doped SWSiCNT, while fixing the tube length and varying the pressure at 293 K. We then plotted the adsorbed hydrogen [loading H 2 (mol/m 3 )] against pressure and the results can be seen in Figs. S1-A to D. [It should be noted that the figures starting with letter S throughout the paper are in “Supplementary Material”]. From Figs. S1-A to E, we can find the following:
From Figure S1-E, we clearly find that SWSiCNT with doping has more hydrogen adsorptivity than SWSiCNT without doping.
The hydrogen loading inside the Li- and K doping increases as a function of pressure.
The behavior and amount of hydrogen adsorption in sites 2, 3 and 5 are similar in both Li- and K-doped SWSiCNT. Li- and K doping in these sites has minimum hydrogen physisorption, although the amount of hydrogen adsorption in these sites in both Li- and K-doped SWSiCNT correspond to the data in the figures and are almost the same and in the range of 1,000 mol/m 3 (low pressure) to 8,000 mol/m 3 (high pressure). In K-doped SWSiCNT, however, hydrogen adsorption is slightly more than that in Li-doped SWSiCNT.
The highest to lowest values of hydrogen adsorption in all of the sites, Li- and K-doped SWSiCNT are as follows: site 1 > site 7 > site 4 > site 6. It should be noted that the hydrogen adsorption in sites 7 and 4 is very close and the results of hydrogen adsorption in sites of 2, 3 and 5 are very close too and lower than other sites. In K-doped SWSiCNT in situations 7f and 4f (Figure S1-C), the hydrogen adsorption is almost equal. This indicates that K doping on (7f and 4f) and (7h and 4h) are almost equal; on the other hand in Li-doping, the doping sites show a slight difference.
Another indication is the lower hydrogen physisorption in sites with the symbol h compared to sites with symbol f. This means that with half the concentration of Li or K as the doping atom, we can achieve lower hydrogen adsorption compared to when all sites are doped with Li or K atoms. Therefore, we can find that hydrogen adsorption in Figure S1-A is more than that in Figure S1-B; this behavior is also observable in Figure S1-C where hydrogen adsorption is more than that in Figure S1-D.
Figure S1-E shows a comparison between the highest hydrogen adsorptivity in Figs. S1-A to D. K-doped SWSiCNT on top of all Si and C atoms and in the middle of the Si–C bonds (K doped on site 1f) has the most hydrogen adsorption in all cases and K-doped SWSiCNT on top of half of the Si and C atoms and in the middle of the Si–C bonds (K-doped on site 1h) is lower than K-doped on site 1f. K doped on site 1h has more hydrogen adsorptivity than Li doped on site 1f. Finally, Li doped on site 1h has the least hydrogen adsorption. To sum up, the highest hydrogen adsorptivity from Figs. S1-A to D are as follows:
K doped on site 1f > K doped on site 1h > Li-doped on site 1f > Li-doped on site 1h.
The last investigation revealed that when the pressure was fixed in Figs. S1-A to D, for instance, at 3 and 9 MPa, there was no significant difference between hydrogen adsorption in all doping sites in Figure S1-A. Also, there was no significant difference between the hydrogen adsorption in all doping sites at 9 and 15 MPa (or 3 and 15 MPa) in Figure S1-A. This is also visible in Figs. S1-B to D. This means, when the pressure increase, a larger amount of hydrogen will be adsorbed, while the difference between the adsorption doping sites remains constant in all cases. Figure S1-E confirms this better.
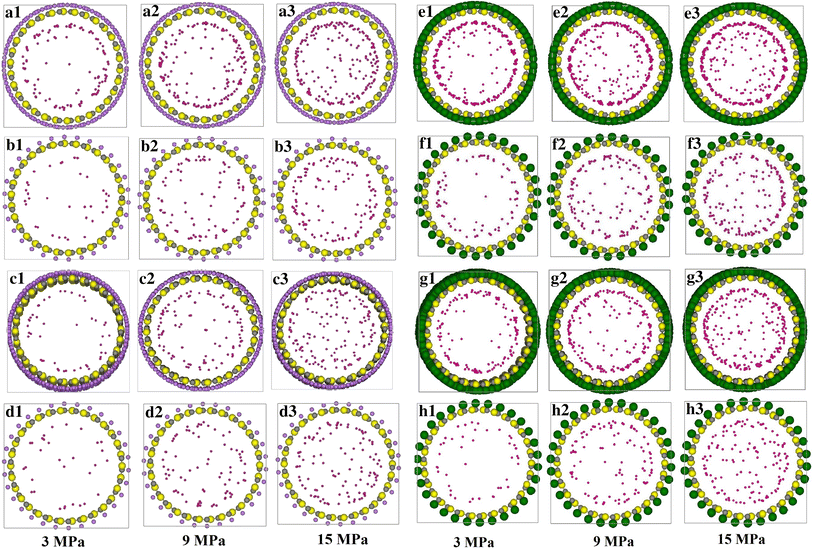
In Fig.
1
a–h, we present the cross sections of Li- and K-doped SWSiCNT with hydrogen adsorbed at 293 K. (for example, a1, a2 and a3 symbols show the hydrogen adsorbed at 293 K in Li- doped on site 1f at 3, 9 and 15 MPa, respectively. Please see figure caption for explanation of other symbols).
a1–a3
Are the cross sections of (15, 15) Li doped on top of all Si, C atoms and bonds of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPam, respectively, at 293 K. These are the maximums of hydrogen physisorption at full positions of Li doping.
b1–b3
Are the cross sections of (15, 15) Li doped on only the top of all Si atoms of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the minimums of hydrogen physisorption at full positions of Li doping. However, Li doped on only the top of all C atoms and Li doped on only the top-middle of the hexagons have the minimum of hydrogen physisorption at full positions of Li-doping.
c1–c3
Are the cross sections of (15, 15) Li doped on top of the half of Si, C atoms and bonds of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the maximum of hydrogen physisorption at half positions of Li doping.
d1–d3
Are the cross sections of (15, 15) Li doped on only the top of the half of Si atoms of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the minimums of hydrogen physisorption at half positions of Li doping. However, Li doped on only the top of all the C atoms and Li doped on only the top–middle of the half of the hexagons have the minimum of hydrogen physisorption at full positions of Li doping.
e1–e3
Are the cross sections of (15, 15) K doped on top of all Si, C atoms and bonds of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the maximums of hydrogen physisorption at full positions of K doping.
f1–f3
Are the cross sections of (15, 15) K doped on only the top of all Si atoms of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the minimums of hydrogen physisorption at full positions of K doping. The K doped on only the top of all C atoms and the K doped on only the top-middle of the hexagons have the minimum of hydrogen physisorption at full positions of K doping.
g1–g3
Are the cross sections of (15, 15) K doped on top of the half of the Si, C atoms and bonds of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the maximums of hydrogen physisorption at half positions of the K doping.
h1–h3
are the cross sections of (15, 15) K doped on only the top of the half of Si atoms of SWSiCNT that have been filled with hydrogen at 3, 9 and 15 MPa, respectively, at 293 K. These are the minimums of hydrogen physisorption at half positions of K doping. However, the K doped on only the top of all C atoms and K doped on only top–middle of the half of hexagons have the minimum of hydrogen physisorption at full positions of K-doping.
Yellow,
gray
,
violet
and
green
atoms are Si, C, Li and K atoms, respectivelyFig. 1
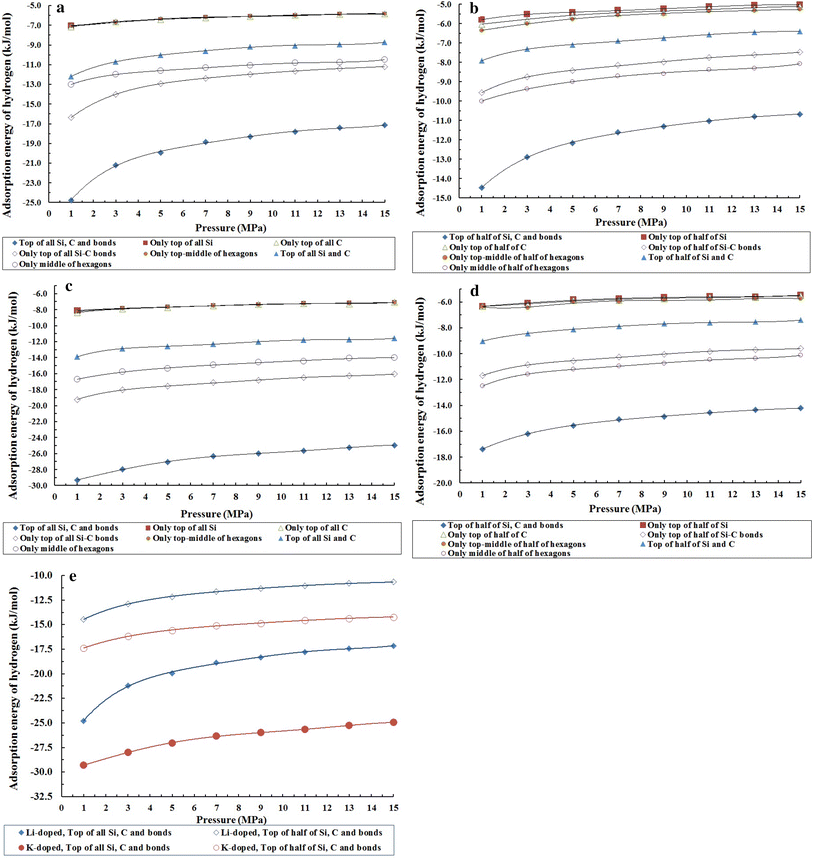
Another investigation is the energy of hydrogen adsorption in Li- and K- doped SWSiCNT in all doping sites. It is clear that the case which has maximum hydrogen adsorption is the maximum absolute value of the adsorption energy. Therefore, it is important to combine the results of Figs. S1-A to D with their hydrogen adsorption energy. Figure
2
a–e presents the plots of the energy of hydrogen adsorption corresponding to Figs. S1-A to E. In Fig.
2
a–e, it is quite clear that the cases with an absolute value of higher energy adsorption are more hydrogen adsorbable. For instance, in Fig.
2
a, the doping site of 1f has minimum energy adsorptivity and this case has the maximum hydrogen physisorption, as shown in Figure S1-A. Furthermore, the sites with lower hydrogen adsorptivity have more energy adsorption. For example, sites 2f, 3f and 5f or the sites 2h, 3h and 5h with the minimum hydrogen adsorptivity have the maximum energy adsorption. There is however one undeniable problem in all investigations in Fig.
2
a–e. At low pressure, the hydrogen adsorption is also low, and when the pressure increases the hydrogen adsorption increases as well. Therefore, at low pressure, the energy adsorption must be more than that of high pressure, because at high pressure the hydrogen adsorptivity will increase and consequently hydrogen energy adsorbed will be accompanied with an increase in absolute value. Therefore, in Fig.
2
a–e, we see that the energy of adsorption is a decreasing function of pressure, while in Fig.
2
a–e, we see the opposite taking place. In Fig.
2
a–d, we observe that the energy adsorptivity is a direct function of pressure. Of course, we expect that the energy adsorptivity is the inverse function of pressure. In fact, when pressure increases, the hydrogen physisorption also increases, in spite of the adsorbent space remaining constant (the interior space of the tube does not change). When increasing the hydrogen storages (loading), the repulsion energy between hydrogen molecules and hydrogen molecules with atoms of the nanotube increases at high pressure, which causes an increase in the adsorption interaction energy. Thus, in Fig.
2
a–d we can observe that energy adsorption is a direct function of pressure. It is due to this concept that the interior space of the tube is an important factor in hydrogen physisorption.
a
Energies of hydrogen adsorption for situations of
Figure S1-A
that have been plotted against pressure at 293 K. The lines correspond to fifth-order polynomial fits in all sub-Figures. 2.
B
Energies of hydrogen adsorption for conditions
Figure S1-B
that have been plotted against pressure at 293 K.
c
Energies of hydrogen adsorption for conditions
Figure S1-C
that have been plotted against pressure at 293 K.
d
Energies of hydrogen adsorption for conditions in
Figure S1-D
that have been plotted against pressure at 293 K.
e
Energies of hydrogen adsorption for conditions in
Figure S1-E
that have been plotted against pressure at 293 KFig. 2
To find another factor of investigation, we calculated the Langmuir and Langmuir–Freundlich equation parameters for hydrogen adsorption by applying the following equations [ 4 , 5 ], respectively [ 10 , 17 ]: where θ is the fraction of adsorbent’s surface (Li- and K-doped SWSiCNT) covered by the adsorbate (hydrogen); P is the partial pressure of hydrogen; and Z max (moles/m 3 ) is the maximum hydrogen loading (corresponding to the complete coverage of the surface of the Li- and K-doped SWSiCNT by hydrogen), while K (in (kPa) −1 ), n and A (the size of which is related to the value of n and is in the order of (kPa) −1 ) are constants).
In this work, we investigated the adsorption of hydrogen inside Li- and K-doped SWSiCNT mathematically by comparing the results obtained by using the Langmuir (L) and the Langmuir–Freundlich (L-F) equations. These equations help us find the best fittings to the hydrogen physisorption data to illustrate the mathematical form of adsorption and the mechanism of hydrogen physisorption (by comparing the equations). The results of Figure S1-A are fitted to the Langmuir and the L-F equations to prepare Fig. S2-A and B, respectively. Indeed, Figure S2-C (Langmuir equation fitted) and D (L-F equation fitted) were prepared corresponding to the results of Figure S1-B, and the Langmuir and L-F equations fitted in Fig. S2- E, F, G and H were achieved from Fig. S1-C and D data. In these diagrams, it can be seen that Fig. S2-A, C, E and G are related to the Langmuir equation fitted to the sorption data. In these figures, we can also see that the Langmuir equation could not properly fit to the adsorption data, while according to Fig. S2-B, D, F and H we can find that the L-F equation fitted well to the adsorption data. The Langmuir and L-F equations correspond to monolayer gas sorption and multi-layer gas sorption in nanotubes, respectively, and the grand canonical ensemble helps us to achieve this goal [ 17 ]. According to previous results, the adsorption of the second layer of gas inside a nanotube with a diameter of <2 nm is thermodynamically unstable [ 10 , 17 ]. It should be noted that the diameter of (15, 15) SWSiCNT is 26.786 Å which is larger than 2 nm, so we expect that the gas sorption inside this nanotube takes place at least with the second-layer mechanism. Fortunately, this quality can be observed in Fig. 2 . Figure 1 illustrate that at low pressure (3 MPa) the molecular hydrogen was to be adsorbed inside the nanotube with monolayer adsorptivity, while Li or K are doped to (15, 15) SWSiCNT. The ability of this nanotube to adsorb hydrogen is increased at 3 MPa, and so the second-layer mechanism of adsorption can be seen (See the Fig. 1 : series a1, b1, c1, d1, e1, f1, g1 and h1 are related to Li- and K-doped SWSiCNT at 3 MPa). The second-layer mechanism of hydrogen adsorption is even more important at high pressures, such as 9 and 15 MPa. At moderate pressure such as 9 MPa, second-layer hydrogen adsorptivity is quite apparent, while at high pressure such as 15 MPa multi-layer gas sorption occurs. Therefore, L-F equation was a better fit to the data at high pressures in the nanotube without doping, and L-F equation was a better fit to the data at moderate and high pressure in nanotube and this indicates that hydrogen adsorptivity occurs in multi-layer gas sorption. In fact, Figs. S2-B, D, F and H emphasize that in Li- and K-doped SWSiCNT multi-layer gas sorption is the dominant mechanism.
After the discussion about hydrogen physisorption inside Li- and K-doped SWSiCNT, we performed a comparison of our data values obtained from our calculations and the data from other works [ 23 , 24 , 26 ]. Figs. S3-A to D represent our data and data from Ref. [ 24 ]. We noted here that our data in Figs. S3-A to D are similar to those in Figs. S1-A to D. In Figure S3-A, we find that site 1f in Li-doped SWSiCNT has more hydrogen adsorptivity than Li- and K-doped single-walled carbon nanotubes’ (SWCNT) bundles. However, other sites in Li-doped SWSiCNT do not potentially have hydrogen adsorption more than Li- and K-doped SWCNT bundles. Of course, this view in Figure S3-B is almost true, except that in Li- and K-doped SWCNT bundles, especially, in Li-doped SWCNT bundles, hydrogen physisorption is very close to the site 1f adsorptivity in Li-doped SWSiCNT. The researchers in Ref [ 24 ] have concluded that the hydrogen adsorption in Li-dope SWCNT bundles is more than in K-doped SWCNT bundles. Our results show that K-doped SWSiCNT has more adsorbable than Li-doped SWSiCNT. This comparison may be incorrect, because the adsorbents are different. Our results indicate that for each specific type of nanotube, a particular atom can act more effectively in the doping process and Li is better for CNT and K for SiCNT. From Figure S3-C we can see that hydrogen adsorption in K-doped SWSiCNT is more than in K- and Li-doped SWCNT bundles spatially, in sites 1f, 4f, 6f and 7f. However, hydrogen adsorption in K-doped SWSiCNT in sites 2f, 3f and 5f has almost the same results with K- and Li-doped SWCNT bundles at about 5 MPa. After 5 MPa, hydrogen adsorption is lower in K- and Li-doped SWCNT bundles. This clearly shows that the best doping to hydrogen adsorption in SWSiCNT takes place when K is the dopant atom on all C and Si and bonds. Furthermore, when the concentration of K decreased, K-doped SWSiCNT lost their hydrogen adsorption capacity. Figure S3-D presents this result. Figure S3-D illustrates that except site 1h that has more hydrogen adsorptivity, sites 4h, 6h and 7h have hydrogen adsorption behavior similar to K- and Li-doped SWCNT bundles at moderate pressure, while at low pressure these half K-doped sites have a little more hydrogen adsorptivity than the K- and Li-doped SWCNT bundles.
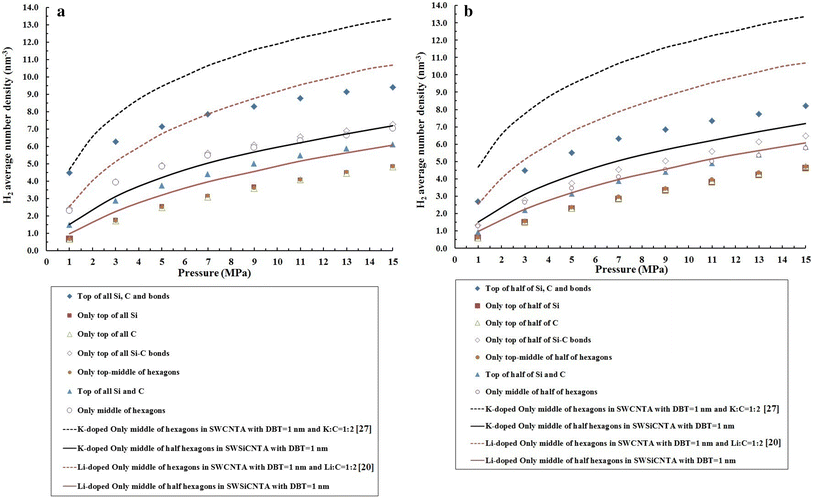
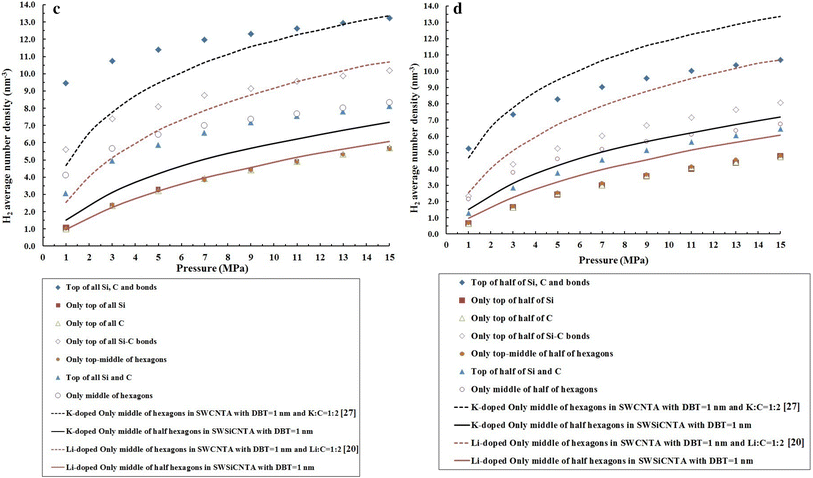
To complete our investigations, we simulated the hydrogen adsorption in K- and Li doping only to the middle of the half of the hexagons in four SWSiCNT which formed arrays with DBT = 1 nm. Figure
3
a–d confirm our results and the results of Refs. [
20
,
26
]. It should be noted that Refs. [
20
,
26
] have investigated K- and Li doping only to the middle of the half of the hexagons in SWCNTA with DBT = 1 nm, while K atoms to C ratio and Li atoms to C ratio are 1–2. Our obtained results and Ref. [
24
] are the opposite of Refs. [
20
,
26
]. According to Figs. S3-A and B, hydrogen adsorption in site 1f and 1h in Li-doped SWSiCNT is more than that in K- and Li-doped SWCNT bundles, while Fig.
3
a, b shows that hydrogen adsorption in K- and Li doping only to the middle of the half of the hexagons in SWCNTA is clearly more than in Li-doped sites f and h of SWSiCNT. This means that the researchers in Refs. [
20
,
26
] have shown that K- and Li doped only to the middle of the half of the hexagons in SWCNTA has more hydrogen adsorptivity than Li-doped SWSiCNT which is opposite to the results of Fig. S3-A and B. It should be noted that in Figs. S3-A and B, K and Li (of course, separately) have been doped at full sites in SWCNT bundles, while in Figs.
3
a, b SWCNTA has been doped by K and Li separately, only to the middle of the half of the hexagons. Although our results and the results of Ref. [
24
] are in agreement, the results of Ref [
20
,
26
] are different. It should be noted that we have performed our simulations using the multipurpose simulation code of Snurr’s research group at the Northwestern University [
28
]. Many articles have already been published that have made use of this simulation package [
10
,
17
,
30
–
32
]. However, we have seen that the results of the Cheng group are slightly larger than our results reported in our previous work [
10
]. In Fig. 6-C and D, the results indicate that K- and Li doped only to the middle of the half of the hexagons in SWCNTA are more than K doped only to the middle of the half of the hexagons in SWSiCNTA. Therefore, we decided to simulate the hydrogen adsorption in K- and Li doped only to the middle of the half of the hexagons in SWSiCNTA to compare these different results. In Fig. 6-A to D, we achieved hydrogen adsorption in K- and Li doped only to the middle of the half of the hexagons in SWSiCNTA. According to Fig.
3
a–d, hydrogen adsorption in K doped only to the middle of the half of the hexagons in SWSiCNTA is more than Li doped only to the middle of the half of the hexagons in SWSiCNTA. Hydrogen adsorption in the following cases is more than K- and Li doped only to the middle of the half of the hexagons in SWSiCNTA: (1) in 1f, 4f and 7f in Li-doped SWSiCNT (See Fig.
3
a); (2) in 1h in Li-doped SWSiCNT (see Fig.
3
b); (3) in 1f, 4f, 6f and 7f in K-doped SWSiCNT (see Fig.
3
c); and (4) in 1h and 4h in K-doped SWSiCNT (see Fig.
3
d). In fact, based on Fig.
3
, the main result is that doping in the nanotube is more important than arraying the nanotube. The doped single-walled nanotubes with alkali atoms have more hydrogen adsorptivity than the alkali atoms-doped single-walled nanotube arrays where alkali atoms have been doped on some sites of the nanotubes.
a
Comparison of adsorption isotherms of hydrogen physisorption based on Li- and K doped only to the middle of the hexagons in SWSiCNTA with DBT = 1 nm (our data) and also based on our data from
Figure. S1-A
and the results in Refs. [
20
,
26
].
B
Comparison of adsorption isotherms of hydrogen physisorption based on Li- and K doped only to the middle of the hexagons in SWSiCNTA with DBT = 1 nm (our data) and also based on our data from
Figure S1-B
and the results in Refs. [
20
,
26
].
c
Comparison of adsorption isotherms of hydrogen physisorption based on Li- and K doped only to the middle of the hexagons in SWSiCNTA with DBT = 1 nm (our data) and also based on our data from
Figure S1-C
and the results in Refs. [
20
,
26
].
D
Comparison of adsorption isotherms of hydrogen physisorption based on Li- and K doped only to the middle of the hexagons in SWSiCNTA with DBT = 1 nm (our data) and also based on our data from
Figure S1-D
and the results in Refs. [
20
,
26
]Fig. 3
Finally, we suggest that K-doped SWSiCNT are better candidates than Li-doped SWSiCNT and SWSiCNT for hydrogen adsorption materials.
In this study, we investigated hydrogen adsorption on seven sites of Li- and K-doped SWSiCNT and SWSiCNTA using GCMC simulation. The results show that hydrogen adsorption in Li- and K-doped SWSiCNT are more than in SWSiCNT except on some doping sites. The observations emphasize that the hydrogen physisorption in Li- and K-doped SWSiCNT is more than in Li- and K-doped SWCNT bundles especially in K-doped SWSICNT. The last consideration is that the Li- and K-doped SWSiCNT to all sites have even more hydrogen adsorptivity than Li- and K-doped SWSiCNTA. Finally, we investigated the mechanism of hydrogen adsorption by fitting the Langmuir and L-F equations to the hydrogen adsorption data and noted that there is a trend toward multi-layer hydrogen adsorptivity in Li- and K-doped SWSiCNT, except for some doping sites where the dominant mechanism is monolayer adsorption of hydrogen.
We acknowledge the University of Zanjan and the Abhar Branch of the Islamic Azad University Research Councils for support to this work.
The authors declare that they have no competing interests.
MS was involved in designing the simulation work, AS wrote the program and ST was involved in getting the data and drafting the manuscript. MS revised the manuscript critically and ZA gave final approval for submission. All authors read and approved the final manuscript.