Department of Chemistry, Payame Noor University, Tehran, 19395-4697, IR
Abstract
In this article, Ru(4,4′-dicarboxy-2,2′-bipyridine)
2
(NCS)
2
dye (N3) and some derivatives were investigated using Density Functional Theory (DFT) calculations in solution to elucidate the influence of the environment and substituted groups on electronic properties. Full geometry optimization and investigation of electronic properties of N3 dye and some derivatives were performed using DFT and HF calculations. The singlet ground state geometries were fully optimized at the B3LYP/3-21G** level of theory through the Gaussian 98 program. Based on the computed results, the optoelectronic properties are sensitive to chemical solvent environments. Moreover, the properties of anatase cluster (TiO
2
) models have been investigated, and N3 dyes have been adsorbed on TiO
2
nano-particle with diprotonated states. The modified N3 dyes highly affected the electronic structure. This leads to significant changes in the adsorption spectra as compared to the N3 dyes. Through hybrid methods, the properties of interfacial electronic coupling of the combined system were estimated. The results of some combined systems showed that the electronic coupling, lowest lowest unoccupied molecular orbitals, and the TiO
2
conduction band resided in the visible region.
Background
Photo-induced phenomenon, based on photon absorption with enough energy, causes the excitation of electrons and then the current generation. Electron transfer (ET) at organic/inorganic interfaces plays a key role in a variety of applications including photocatalysis, photoelectrolysis, and solar cell and nano-scale electronics [
1
–
5
]. The dye-sensitized nano-crystalline solar cell, or Grätzel cell, is a popular alternative in comparison to the costly traditional solar cell [
3
,
6
]. The attached chromophore dye with tuned desired wavelength in a well-known Grätzel cell should absorb photons and generate electrons within dye-sensitized titanium dioxide nano-particles [
7
–
10
]. Quantum chemical calculations can provide information about geometrical and electronic structure in dye-sensitized solar cells (DSSCs) [
11
]. The N3 dye, as shown in Figure
1
, is one conventional dye which can show optical adsorption spectra [
12
] related to the size and conjugation of linkers to the surface at semiconductor TiO
2
. Surface electron transfer accompanying the initial light absorption and excitation of ruthenium dyes often leads to efficient photo-induced charge separation across the dye and TiO
2
interfaces.
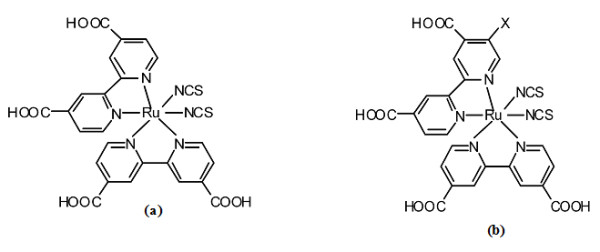
Figure 1
N3 dye (a) and its α substituted derivatives.
The structural formulae of N3 dye (
a
) and its α substituted derivatives (
b
) with
X
= CH
3
, C
2
H
5
, NH
2
, N(CH
3
)
2
, F, and Cl.
TiO
2
can be formed in several phases such as anatase, rutile and brookite. There are some properties of TiO
2
nano-crystals such as electro-opticality, low cost, chemical stability, non-toxicity, abundance, availability, and lack of erosion and corrosion against light which are common in the literature [
13
]. The anatase crystal phase of TiO
2
has an octahedral structure with wide gap energy, photo-induced activity, large surface area-to-bulk ratio, and can absorb UV light more than other phases. Due to the lack of anatase stability in its structure and a high degree of dangling bonds, calculation of structural and electronic properties of the large number of under coordinated atom is complicated. However, the size and complexity of TiO
2
nano-particles impose challenges for choosing the methods. Several titanium oxide nano-crystals have been investigated in some studies [
14
–
19
]. The calculation for anatase (TiO
2
)
5
and (TiO
2
)
16
clusters has been performed by DFT, but because of computational demand for large systems, the semi-empirical methods have been used. The (TiO
2
)
54
cluster has large surface and has more tendency toward sphericity, and it is selected as the basis for computing absorption dyes and its derivatives.
The Ru(4,4′-dicarboxy-2,2′-bipyridine)
2
(NCS)
2
or ‘N3’ dye and its derivatives are probably the most well known sensitized dyes which transfer electron by adsorbing sunlight followed by electron injection to the conduction band of TiO
2
, so the gap energy will be increased [
20
,
21
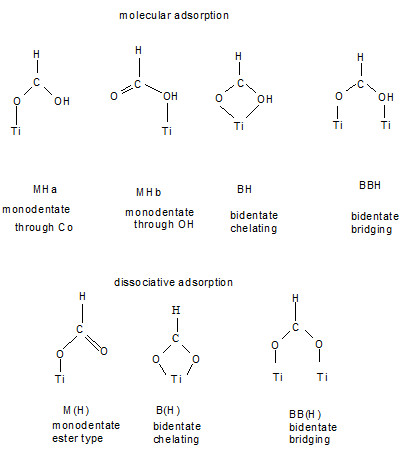
]. The electron transfer depends on different parameters, for example, the type of attachment such as bridge, chelating, mono-dentate which are shown in Figure
2
[
22
,
23
]. The dye is adsorbed in a prototype binding of the two carboxylic acid anchor groups of one of the bipyridine ligands in a bridging bi-dentate adsorption mode. Figure
2
shows the types of attachment anchor groups on the adsorbent layer [
4
].
Figure 2
The types of attaching anchor groups on the adsorbent layer
[
4
].
The purpose of this paper is to investigate the role of acceptor and donor groups on N3 dyes and to understand the electron transfer phenomena, variation of gap energy, and the geometrical and electronic coupling among the parts in the combined system, (TiO
2
)
54
-N3 derivatives.
Methods
Through Density Function Theory (DFT), the full geometry optimization and investigation of the electronic properties of N3 dye and some derivatives were performed [
11
]. The results were obtained by DFT and supported by Hartree-Fock (HF) calculations. The singlet ground state geometries were fully optimized at the B3LYP/3-21G** level of theory. All calculations were performed with Gaussian 98 program (Gaussian, Inc., Wallingford, CT, USA) [
24
].
The variation of gap energies was investigated by inserting electron donor groups such as CH
3
, C
2
H
5
, NH
2
, N(CH
3
)
2
, or electron accepter groups such as F and Cl on the α position related to carboxylate on N3 dye as shown in Figure
1
. To compare of the results, all calculations were done first in gas phase and then in solvent phase. In this study, PCM model of ethanol (dielectric constant,
ϵ
= 25) and acetonitrile (
ϵ
= 37) was used.
Since the DFT calculation of the large clusters is very costly, the (TiO
2
)
54
nano-crystals have been optimized by MSINDO semi-empirical method [
3
]. Further, we used the integrate B3LYP:ZINDO1 approach for the combined dyes and (TiO
2
)
54
cluster systems and some of the dye derivates.
To evaluate the accuracy of the proposed hybrid method results, the combined system of N3 and (TiO
2
)
54
has been optimized via the DFT method by a 16-core supercomputer. The duration of the each calculation was about 2 months.
Results and discussion
The computational results for the N3 dyes and some derivatives which were performed by B3LYP method and 3-21G** basis set in both gas and solvent phases are presented in Table
1
. A visual comparison of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of N3 and some of its derivatives in the gas, acetonitrile, and ethanol phases shows an increasing and obvious shifting in absolute energy and reveal a clear difference in the energetic ordering of the frontier orbitals in different environments.
Table 1
The calculated HOMO, LUMO (in Hartree), gap energy, and adsorption threshold by DFT
Gas phase
Ethanol
Acetonitrile
-X
HOMO
LUMO
Gap (V)
λ(nm)
HOMO
LUMO
Gap (eV)
λ(nm)
HOMO
LUMO
Gap (eV)
λ(nm)
-H
−0.1063
0.1085
1.494
830
−0.1976
−0.1202
2.105
620
−0.1988
−0.1203
2.136
581
-CH3
−0.1726
−0.1172
1.508
822
−0.1943
−0.1178
2.082
595
−0.1955
−0.1179
2.109
588
-C2H5
−0.1627
−0.1052
1.565
792
−0.1867
−0.1164
1.913
648
−0.1910
−0.1172
2.008
614
-NH2
−0.1696
−0.1117
1.574
788
−0.1872
−0.1145
1.978
626
−0.1881
−0.1147
1.996
621
-N(CH3)2
−0.1613
−0.1022
1.609
770
-
-
-
-
−0.1592
−0.1055
1.462
848
-F
−0.1741
−0.1191
1.498
828
−0.1942
−0.1216
1.977
627
−0.1980
−0.1223
2.059
602
-Cl
−0.1741
−0.1197
1.479
838
−0.1928
−0.1232
1.894
654
−0.1981
−0.1243
2.008
618
The results show that the substituted groups do not have an important role in transferring the absorption wavelength from UV to visible region in the gas phase. Moreover, the groups have a few influences on the decrease of N3 gap energy.
The energy silently decreases in solvent environment, so the difference between gas and solvent environment is about 0.5 eV for all groups. The solvent effect on the gap energy variation occurs through the interaction of the dye and ethanol or acetonitrile molecules. It has a significant influence on the optical properties of these complexes by the environment, so the wavelength of N3 complexes is shifted from 830 to 580 nm by inclusion of the solvent. The range of variation of optoelectronic properties is taken into account under a model for complexes with conjugated properties whether or not the theoretical modeling is capable of providing a physically realistic description of the complexes with extended conjugation.
Using the integrate B3LYP:ZINDO1 approach, the results of the combined systems of N3 dyes and some of the derivatives with (TiO
2
)
54
cluster are summarized in Table
2
. The table shows that some of the absorption wavelengths of a combined system are located in the visible area. Furthermore, the computed results by the mentioned methods for heterogeneous super molecule are given in Table
3
. However, the gap energy almost differs between ONIOM and DFT; ONIOM results are in agreement with our expectation.
Table 2
The calculated gap energy and adsorption threshold by ZINDO1 of theory
Molecules
Gap (eV)
λ(nm)
(TiO2)54
3.97
313
(TiO2)54 + N3
1.54
800
(TiO2)54 + N3 + F
4.21
295
(TiO2)54 + N3 + Cl
6.6
93
(TiO2)54 + N3 + CH3
2.7
445
(TiO2)54 + N3 + C2H5
2.5
490
(TiO2)54 + N3 + NH2
1.6
780
(TiO2)54 + N3 + N(CH3)2
0.3
4,353
Table 3
The calculated gap energy and adsorption wavelength of combined system of N3 and (TiO2)54cluster
Method
Gap (eV)
λ(nm)
ONIOM
1.54
800
B3LYP
3.93
320
Conclusion
To significantly develop DSSCs and related photo-electrochemical devices, the quantum chemical calculations were performed to provide a better theoretical understanding of the basic physical and chemical processes of dye-sensitized semiconductors. As shown before, the modified N3 dyes have highly affected the electronic structure. This leads to significant changes in the absorption spectra as compared to the N3 dyes. The optical absorption spectra are related to the environment, but not probably related to the type of
X
-substituted groups. The results of the combination system showed that the electronic coupling of the lowest dye LUMOs and the TiO
2
conduction band is negligible. Moreover, the lowest LUMO dye and TiO
2
band conduction, using hybrid method and the electronic structure, agree with the experiment evidence. Finally, the absorption wavelength of some substituted dyes of combined system is located in the visible area.
Acknowledgment
The authors greatly appreciate Dr. HS Wahab from the Baghdad University of Technology for his instructions.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MO participated in the design and the sequence alignment of the study. LT carried out the jobs by Gaussian and participated in the evaluation of the data and in drafting the manuscript. Both authors read and approved the final manuscript.
References
Ferrere and Gregg (2001) Large increases in photocurrents and solar conversion efficiencies by UV illumination of dye sensitized solar cells (pp. 7602-7605) 10.1021/jp011612o
Derosa and Seminario (2001) Electron transport through single molecules: scattering treatment using density functional and Green function theories (pp. 471-481) 10.1021/jp003033+
O'Regan and Grätzel (1991) A low-cost, high-efficiency solar cell based on dye sensitized colloidal TiO2 films (pp. 737-740) 10.1038/353737a0
Pan et al. (2002) Photoinduced electron transfer between a carotenoid and TiO2 nanoparticle (pp. 13949-13957) 10.1021/ja0279186
Biju et al. (2004) Intermittent single-molecule interfacial electron transfer dynamics (pp. 9374-9381) 10.1021/ja040057b
Rego and Batista (2003) Quantum dynamics simulations of interfacial electron transfer in sensitized TiO2 semiconductors (pp. 7989-7997) 10.1021/ja0346330
Stier et al. (2003) Ab initio molecular dynamics of ultrafast electron injection from molecular donors to the TiO2 acceptor (pp. 132-146) 10.1117/12.503646
Duncan et al. (2005) Ab initio nonadiabatic molecular dynamics of the ultrafast electron injection across the alizarin-TiO2 interface (pp. 7941-7951) 10.1021/ja042156v
Lundqvist (2006) Uppsala University
Aghtar (2009) Payame Noor University (PNU)
Diebold (2003) Structure and properties of TiO2 surfaces: a brief review (pp. 681-687) 10.1007/s00339-002-2004-5
Nilsing et al. (2005) Phosphonic acid adsorption at the TiO2 anatase (101) surface investigated by periodic hybrid HF-DFT computations (pp. 49-60) 10.1016/j.susc.2005.02.044
Homann et al. (2004) Adsorption of small molecules on the anatase (100) surface (pp. 135-144) 10.1016/j.susc.2003.12.039
Vittadini et al. (2000) Formic acid adsorption on dry and hydrated TiO2 anatase (101) surfaces by DFT calculations (pp. 1300-1306) 10.1021/jp993583b
Nilsing et al. (2005) Anchor group influence on molecule-metal oxide interfaces: periodic hybrid DFT study of pyridine bound to TiO2 via carboxylic and phosphonic acid (pp. 375-380) 10.1016/j.cplett.2005.08.154
Rappoport et al. (2009) Approximate density functionals: which should I choose? (pp. 159-172) Wiley
Nazeeruddin et al. (2005) Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers (pp. 16835-16847) 10.1021/ja052467l
Wahab et al. (2008) Computational modeling of the adsorption and photodegradation of 4-chlorophenol on anatase TiO2 particles (pp. 84-90) 10.1016/j.theochem.2008.05.019
Lundqvist et al. (2006) DFT Modeling of bare and dye-sensitized TiO2 nanocrystals (pp. 3214-3234) 10.1002/qua.21088
Lundqvist et al. (2007) Calculated optoelectronic properties of ruthenium tris-bipyridine dyes containing oligophenyleneethynylene rigid rod linkers in different chemical environments (pp. 1487-1497) 10.1021/jp064219x