Results and discussion
Armchair (3,3) and zigzag (5,0) ultra-narrow boron nitride nanotubes terminated with hydrogen atoms, each having 15 meshes, have been selected for the present study. Both of these have different chirality but are diametrically almost equal and both of these are wide-band-gap semiconductors. Figure
1a,b
shows the AM1-optimized structures obtained after one K atom was relaxed in the neighborhood of the already optimized structure of the pristine (3,3) and (5,0) BNNTs, respectively. As can be seen from these figures, AM1 force-field optimization resulted in the adsorption of the K atom on the surface of the exterior wall of the optimized structures of (3,3) and (5,0) BNNTs. The AM1-optimized bond lengths in the mesh near the dopant K atom before and after doping potassium atom on the (3,3) and (5,0) BNNTs are listed in Table
1
from which a distortion of BNNTs after potassium doping can be observed. The same is also apparent in Figure
1a,b
from which it can be seen that the K atom occupies the hollow site on one of the hexagons of the BNNT. Adsorption energy of K atom doped on BNNT is evaluated according to the expression:
where
E
(NT + K) is energy of K-doped BNNT,
E
(NT) is energy of nanotube, and
E
(K) is free energy of potassium atom.
AM1-optimized structure of K(yellow)-decorated (a) (3,3) and (b) (5,0) BNNTs.
Red ball for nitrogen and blue for boron atom.Figure 1

Table 1
Changes of bond length of the associated mesh of BNNTs after doping and adsorption of potassium
(3,3) BNNT | (5,0) BNNT | ||||
|---|---|---|---|---|---|
B5-B13 | 1.4653 | 1.5064 | B25-N42 | 1.4655 | 1.5045 |
B5-N6 | 1.4601 | 1.4435 | B25-N22 | 1.4437 | 1.4321 |
B19-N6 | 1.4596 | 1.5927 | B23-N22 | 1.4675 | 1.5398 |
B19-N23 | 1.4523 | 1.5100 | B23-N2 | 1.4675 | 1.5398 |
B22-N13 | 1.4432 | 1.4831 | B32-N42 | 1.4655 | 1.5045 |
B22-N23 | 1.4690 | 1.4510 | B32-N2 | 1.4675 | 1.4321 |
The process of adsorbing K atom on BNNTs is found to be exothermic with binding energy of 3.2 and 2.8 eV/K atom for (3,3) and (5,0) BNNTs, respectively. Margine and Crespi[
36
] reported that the presence of strong electron donor alkali metals on BNNTs provided positive potential surrounding alkali atoms. The calculated dipole moment of 9.5 D and 9.7 D for (3,3) and (5,0) K-doped BNNTs, respectively, is relatively large because of the presence of potassium atom that is donating the positive charge to the BNNT. The large enhancement in the calculated value of the dipole moment of the BNNT has also been predicted using advance level of theory by Farmanzabeh and Ghazanfary[
37
]. The same is also supported by Mulliken charge analysis which revealed a charge transfer of 0.697
e
−
from decorated potassium atom to (3,3) BNNT and 0.71
e
−
to (5,0) BNNT, respectively. The redistribution of charge within BNNT is markedly over the mesh which is nearest to potassium atom. The boron constituent still has positive charge which has been slightly reduced, while negative charge on nitrogen has increased a little. The main contributions to the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) in K-doped BNNTs are from nearest neighboring atoms of potassium and have feeble participation of
s
and
p
orbitals from potassium side; on the other hand, for pristine BNNTs, the HOMO is largely consisted of
p
orbital of constituent nitrogen atoms, and LUMO consists of
s
orbital of boron atoms. In order to examine the effect of K doping on the electronic structure of BNNTs, the PDOS of the doped BNNTs are plotted and are shown in Figure
2
. It is the presence of potassium atom due to which the contribution to inter-frontier orbitals by the labeled constituents got manifested. It is the alpha DOS of decorated sample that has the change in electronic state. The conductivity of BNNTs gets enhanced due to the appearance of a peak on the top of the valence band in the presence of potassium adsorbate.
PDOS for K-doped (3,3) (a) and (5,0) (b) BNNTs are plotted.
Color on line represents the various fragments.Figure 2

Adsorption of a single H2
The H
2
molecule can be adsorbed to the K-decorated BNNTs through K atom with distance of 2.5 and 2.49 Å from K atom in (3,3) and (5,0) K-decorated BNNTs, respectively. The two H-K bond lengths are 2.52 and 2.54 Å for both the (3,3) and (5,0) BNNTs; however, there is slight elongation in H-H bond length in (5,0) BNNT. Binding-energy value is 0.53 eV/H
2
molecule for K-doped (3,3) BNNT while in the case of (5,0) nanotube it is slightly less, 0.51 eV/H
2
molecule. The predicted adsorption energy is defined as:
where n is the number of free hydrogen molecule.
The orientation of hydrogen molecule is side-on with respect to K atom in both the nanotubes as can be seen in Figure
3
. Figure
4
shows the PDOS of the single H
2
molecule adsorbed on the (3,3) and (5,0) K-decorated BNNTs. The PDOS analysis, by considering H
2
molecule, potassium atom, and BNNT as individual fragments, revealed that it is the hybridization of the empty K-4
p
orbitals with H
2
σ
orbital that contributes to the H
2
adsorption for both (3,3) and (5,0) nanotubes. The charge decomposition analysis considering K atom and a single H
2
molecule as fragment-1 and (3,3) BNNT as fragment-2 revealed that there is a net charge donation of 0.847
e
−
for alpha MOs from fragment-1 to fragment-2 in case of alpha DOS; for beta DOS, there is a charge donation of 0.156
e
−
from fragment-2 to fragment-1. Similarly, for K-doped (5,0) BNNT, a net charge donation of 0.853
e
−
from fragment-1 to fragment-2 takes place for alpha DOS, whereas a net charge donation of 0.148
e
−
is predicted from fragment-2 to fragment-1 in case of beta MOs. These donations are net result of donation from fragment-1 to fragment-2 and back-donation from fragment-2 to fragment-1. The self-polarization of fragment orbital plays almost negligible role in charge transfer. The Mullikan population analysis revealed that the hydrogen molecule has unequal charges (
viz
0.050
e
−
and 0.029
e
−
) on its two atomic centers, and it is slightly polar in nature as a result of which it seeks K atom which carry 0.612
e
−
charge over it. It is suggested that the charge transfer leaves the metal dopant in cationic form so that the hydrogen molecule can be attracted by the metal cation via the charge polarization mechanism.
H
2
molecule adsorbed on potassium-decorated (3,3) and (5,0) BNNTs. PDOS for single H
2+
K-decorated (3,3) BNNT.
Color online are fragment-wise selection, (
a
) for (3,3) and (
b
) for (5,0).Figure 3
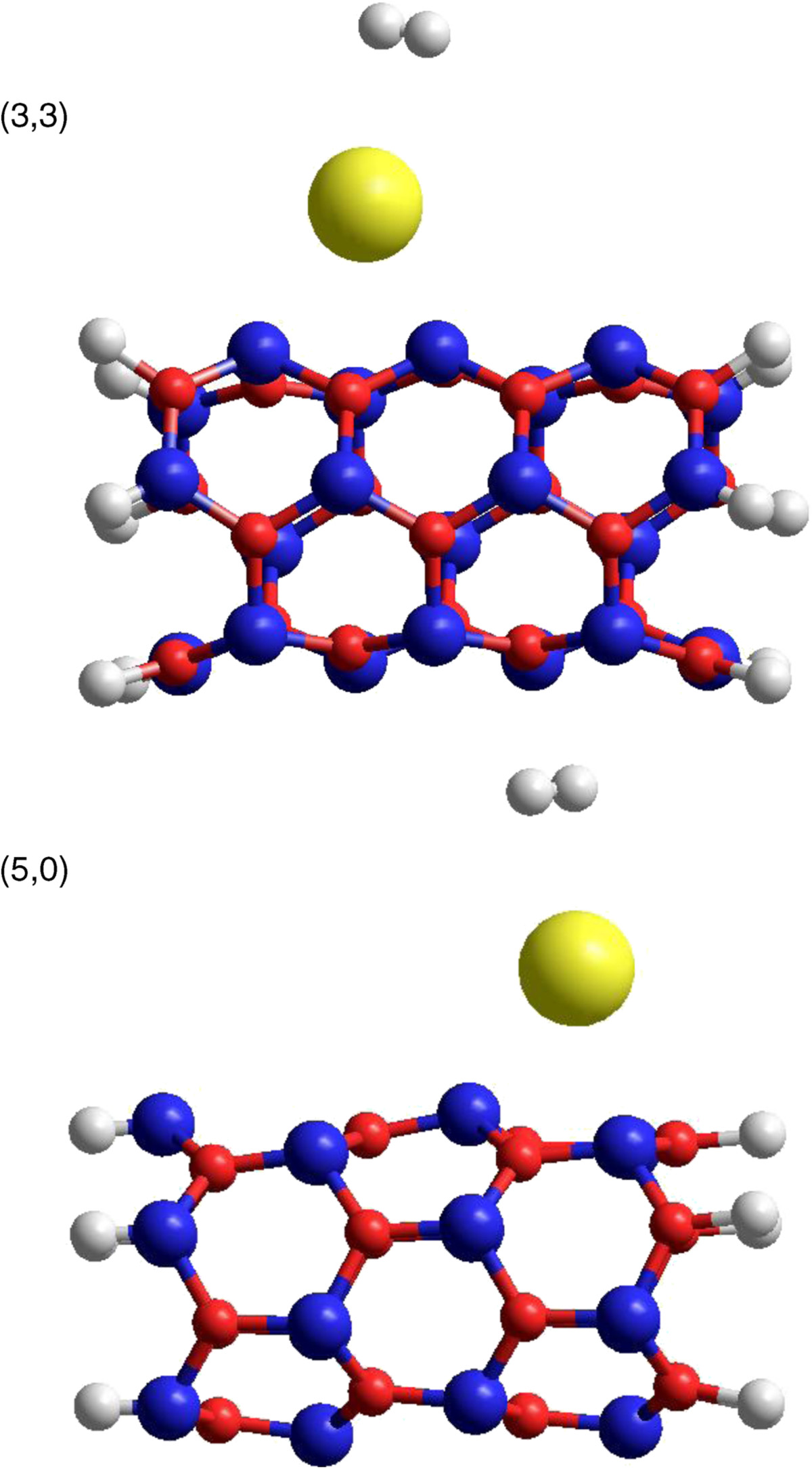
Figure 4

Adsorption of multiple H2
The adsorption of the second H
2
molecule resulted in a decrease in binding energy per H
2
molecule for both (3,3) and (5,0) K-doped BNNTs with values 0.49 and 0.51 eV/H
2
, respectively. There is almost no change in bond length of H
2
molecules. Mulliken population analysis has shown that it is only the redistribution of charges between K atom and H
2
molecules which is responsible for binding of the second H
2
molecule. The charge configuration on the surface of BNNTs is affected negligibly by adsorption of the second hydrogen molecules. The K atom is found to have a slightly reduced charge of 0.544
e
−
. On the other hand, the hydrogen molecules are observed to have unequal distribution of charge within its hydrogen atoms with values of 0.058
e
−
and 0.018
e
−
for the first H
2
and 0.059
e
−
and 0.014
e
−
on the second H
2
. On progressively adding more hydrogen molecules, the charge on K atom is seen to decrease. The charge on K atom is found to be 0.339
e
−
for the adsorption of eight H
2
molecules. The optimization calculations for different numbers of H
2
molecules for the adsorption on K-doped BNNTs were carried out, and the calculated values of binding energy per hydrogen are presented in Table
2
. It is found that one K atom can at most adsorb eight H
2
molecules with
E
b
of 0.38 and 0.41 eV/H
2
for (3,3) and (5,0) BNNTs, respectively. Multiple hydrogen molecules adsorbed on K-doped BNNTs are shown in Figure
5
. These results indicate that the binding energy of H
2
has little to do with the chirality of BNNTs.
Binding energy per hydrogen molecule (in eV) on potassium-decorated BNNT 1H2 2H2 3H2 4H2 5H2 6H2 7H2 8H2 (3,3) K-doped BNNT 0.51 0.49 0.48 0.45 0.43 0.41 0.40 0.38 (5,0) K-doped BNNT 0.53 0.51 0.50 0.48 0.46 0.44 0.42 0.41 Multiple hydrogen adsorption on K-decorated (a) (3,3) and (b) (5,0) BNNTs.Table 2
Figure 5

Hydrogen adsorption on high-coverage K-decorated BNNT
A further study for the stable K adsorbates with high coverage on boron nitride nanotube resulted in a maximum coverage of two fifth on the (3,3) and one third on (5,0) nanotube with
E
b
of 2.3 and 2.8 eV, respectively. For (5,0) BNNT, a maximum coverage of one third with K atoms can be made as clustering of K atoms started with addition of more potassium atoms due to the nearest neighboring effect; as for the stable K adsorbates on boron nitride nanotube, K-K distances should be larger than that of bulk K. Similarly, maximum K converge of two fifth is possible on (3,3) BNNT with 6-K atoms. The (3,3) K-decorated BNNT with maximum coverage of two fifth is found to adsorb at most 36 H
2
with
E
b
of 0.41 eV/H
2
, corresponding to a hydrogen uptake of 8.5 % by weight. For (5,0) K-doped BNNT with maximum coverage, the number of H
2
molecules adsorbed is found to be 37 with
E
b
of 0.55 eV/H
2
with uptake of 9.4 % by weight. The optimized molecular structures of these K-decorated BNNTs with maximum coverage and possible adsorbed hydrogen molecules are shown in Figure
6
. It can be inferred from these results that the zigzag (5,0) K-decorated BNNTs are more favorable for hydrogen adsorption.
Coverage of K atom on (3,3) and (5,0) nanotubes.
Two-fifth coverage of K atom on (3,3) nanotube with 36 H
2
molecules adsorbed and one third coverage on (5,0) nanotube with 37 H
2
molecules.Figure 6
